
A Pruritic Bullous Eruption in a Teenage Boy
A previously healthy 14-year-old boy from Guyana presented to the pediatric emergency room with a 1-week history of worsening pruritic papulovesicular eruption. The eruption began on his arm and spread to his scalp, chest, and legs. He had not used any topical creams or medications, had no significant past medical history, did not take any medications, and met all his developmental milestones appropriately. The family history was unremarkable, and no one in the household had similar symptoms.
Physical examination results revealed a well-developed boy with multiple polymorphous annular tense bullae on his forearms with a negative Nikolsky sign, hundreds of hyperpigmented patches and plaques on his lower extremities, and numerous flesh-colored macules, papules, and excoriations in various stages of healing on his hands, legs, arms, (Figure 1) and chest. Multiple intraoral ulcerations were also noted.
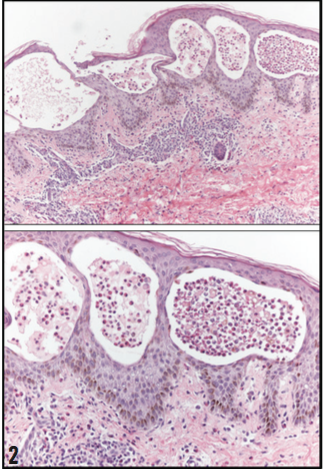
A 4 mm punch biopsy of the abdomen was performed. He- matoxylin and eosin stain revealed extensive vesiculation with eosinophilic infiltrate and focal separation of the epidermis from the dermis (Figure 2). Direct immunofluorescence of a salt-split skin substrate revealed 1+ linear C3 deposition at the basement membrane zone with trace linear immunoglobulin G (IgG) deposition. No immunoglobulin A (IgA) deposition was noted.

What is the cause of this boy’s pruritic bullous eruption?
(Answer and discussion on next page)
Answer: Bullous pemphigoid
The boy was diagnosed with bullous pemphigoid, an autoim- mune subepidermal blistering skin condition that is rarely noted in children and has a median age onset of 75 years.1,2 It occurs equally in males and females and has no predisposition to a par- ticular race.1 The condition is characterized by multiple tense bullae appearing on normal-looking skin or on an erythematous base. Nikolsky sign is usually negative. Upon rupture of the bul- lae, the erosion does not spread beyond the borders as seen in pemphigus vulgaris. Erythematous macules, papules, and ero- sions with crusts can also be seen most commonly distributed over the abdomen, groin, and flexor surfaces of the upper and lower extremities. In up to 40% of cases, the mucus membrane can be involved, but this is not typically the initial symptom.2 Pruritus is a common initial symptom, even months before any skin lesion is present, and the literature reports 6 cases of bullous pemphigoid without the presence of the characteristic bullae.3
Bullous pemphigoid develops due to IgG autoantibodies against bullous pemphigoid (BP) 180 and BP230, which are part of the hemidesmosome junctional complex at the dermal-epider- mal junction.4-6 Subsequent attraction of aggressive B and T cells to the bound IgG causes destruction of the complex and leads to subsequent bullae formation.6
Although the cause of bullous pemphigoid is unknown, history of lichen planus, psoriasis, exposure to radiograph, and exposure to ultraviolet radiation has been reported. Medications includ- ing furosemide, nonsteroidal anti-inflammatory drugs, captopril, penicillamine, and antibiotics have also been implicated. The
generalized bullous form is the most common presentation, with tense bullae that do not break easily. It is found preferably on flex- ural areas of the lower abdomen, inner thighs, or flexor forearms.1
Diagnosis requires a histopathologic analysis with subepider- mal vesiculation and separation of the epidermis from the dermis. The biopsy should be obtained from the border of a fresh lesion if possible. Eosinophilic infiltrates can also be visualized on mi- croscopic examination, but they are not a criterion for diagnosis. An indirect immunofluorescence (IDIF) reveals circulating IgG autoantibodies in 70% to 80% of the patients’ sera that target the basement membrane. Direct immunofluorescence (DIF) re- veals basement membrane deposits of linear IgG in 70% to 90% of patients and C3 in 90% to 100% of patients.1 Lack of IgA deposition usually rules out linear IgA dermatitis. However, IgA deposits have been reported to be present in 25% of cases of bul- lous pemphigoid.7
DIFFERENTIAL DIAGNOSIS
The differential diagnosis for childhood bullous pemphigoid includes bacterial impetigo, herpes simplex, linear IgA dermatosis, epidermolysis bullosa acquisita, dermatitis herpetiformis, pemphi- gus vulgaris, and Stevens-Johnson syndrome. Culturing the exu- date can rule out bacterial and viral causes. Linear IgA dermatosis presents with linear IgA deposits on direct immunofluorescence (DIF) and tense bullae forming on the periphery of previously ruptured bullae, resulting in the unique annular appearance.8,9
This pattern is typically not observed in bullous pemphigoid. Epidermolysis bullosa is a chronic disease with poor response to glucocorticoids. Neutrophils are present in the subepidermal blis-
ters, and C3 deposition is rarely seen. Dermatitis herpetiformis is commonly seen in patients with gluten insensitivity, shows gran- ular IgA deposits on DIF, and is less likely to present with large, tense bullae.1 Pemphigus vulgaris has flaccid bullae with positive Nikolsky sign, and it initially presents with painful oral lesions, with bullae on the skin arising several months later.1,10,11 Finally, Stevens-Johnson Syndrome presents with a high fever followed by painful oral mucosal lesions and most commonly occurs due to medications such as penicillin and anticonvulsants.12,13
Treatment of bullous pemphigoid includes a combination of topical corticosteroid cream such as clobetasol 0.05% and pred- nisolone with initial dose to not exceed 0.75 mg/kg/day.12 Im- munosuppressive agents, including dapsone, azathioprine, and methotrexate have also been previously used.15 Healing of the lesions is usually without scarring, but postinflammatory hyper- pigmentation or hypopigmentation may occur. Our patient was started on a tapered dose of oral corticosteroids, and his lesions resolved on follow-up.
Vesiculobullous diseases in the pediatric population are diffi- cult to differentiate based on clinical findings alone, and a skin bi- opsy can help guide treatment if the diagnosis is unclear. A com- bination of multiple ruptured bullae and inadequate oral intake in a patient with painful oral lesions necessitates close monitoring and proper treatment with topical and systemic corticosteroids. n
Marjon Vatanchi, MD, is a dermatology resident in the De- partment of Dermatology at SUNY Downstate Medical Center in Brooklyn, New York.
Maxim Polansky, MD, is a dermatology resident in the Depart- ment of Dermatology at Emory University in Atlanta, Georgia.
Kevin Wu, MD, is a RESTOR program research fellow at the United States Army Institute of Surgical Research in San Antonio, Texas.
Dakshayani Guttal, MD, is the director of in-patient services in Jamaica Hospital Medical Center and Director of Research in the Residency Program of Pediatrics at Flushing Hospital Medical Center in Jamaica, New York.
REFERENCES
Person JR, Rogers RS 3rd. Bullous and cicatricial pemphigoid. Clinical, histopathologic, and immunopathologic correlations. Mayo Clin Proc. 1977;52(1):54-66.
Korman N. Bullous pemphigoid. J Am Acad Dermatol. 1987;16(5 Pt 1): 907-924.
Alonso-Llamazares J, Rogers RS 3rd, Oursler JR, Calobrisi SD. Bullous pemphigoid presenting as generalized pruritus: observation in six patients. Int J Dermatol. 1998;37(7):508-514.
Labib RS, Anhalt GJ, Patel HP, Mutasim DF, Diaz LA. Molecular heterogeneity of the bullous pemphigoid antigens as detected by immunoblotting. J Immunol. 1986;136(4):1231–1235.
Stanley JR, Tanaka T, Mueller S, Klaus-Kovtun V, Roop D. Isolation of complementary DNA for bullous pemphigoid antigen by use of patients’ autoantibodies. J Clin Invest. 1988;82(6):1864–1870.
Di Zenzo G, Thoma-Uszynski S, Calabresi V, et al. Demonstration of epitope- spreading phenomena in bullous pemphigoid: results of a prospective