
Infant With Recurrent Pneumonia and Reflux: Airway or Foregut Malformation?
Authors:
Deepa D. Kulkarni, MD; Gitanjli Arora, MD; Daniel DeUgarte, MD; and Stephen B. Shew, MD
Dr Kulkarni is a pediatric resident at Mattel Children’s Hospital at the University of California, Los Angeles (UCLA). Dr Arora is a pediatric hospitalist at Mattel Children’s Hospital UCLA and a clinical instructor in the Department of Pediatrics at the David Geffen School of Medicine at UCLA. Dr DeUgarte and Dr Shew are pediatric surgeons in the Department of Surgery and associate professors of surgery at the David Geffen School of Medicine at UCLA.
Citation:
Kulkarni DD, Arora G, DeUgarte DA, Shew SB. Infant with recurrent pneumonia and reflux: airway or foregut malformation? Consultant for Pediatricians. 2013;12(12):544-546.
A 3,345-g boy was born at full term to a primigravid mother via an uncomplicated cesarean delivery for failure to progress. At 19 weeks’ gestation, routine fetal ultrasonography revealed a cystic lesion in the right lung.
Fetal magnetic resonance imaging was performed to further characterize the lesion, and findings were suggestive of extralobar pulmonary sequestration in the right lower lobe.
Postnatally, the patient was monitored in the neonatal intensive care unit, where chest radiography confirmed a cystic mass in the right lower lobe measuring 3 cm in diameter, with an appearance more consistent with congenital pulmonary airway malformation (CPAM) as opposed to pulmonary sequestration. The patient’s respiratory status remained stable, and he was feeding without difficulty. He was discharged uneventfully after a short period of observation.
Between the ages of 10 days and 4 months, the patient was admitted 4 times to various institutions with symptoms of fever, cough, emesis, and respiratory distress. Each time, he was treated for bacterial pneumonia. Repeated imaging studies during these acute illnesses, including chest radiography and computed tomography with intravenous contrast, continued to suggest a right lower-lobe CPAM with likely superimposed recurrent pneumonia.
Between illnesses, the infant was noted to have wheezing and chronic cough, for which he was started on inhaled β-agonist therapy for suspected reactive airway disease. In addition, he had symptoms of emesis attributed to gastroesophageal reflux, for which he was started on proton-pump inhibitor therapy. He otherwise continued to develop and gain weight appropriately.
At 7 months of age, the patient presented again to a hospital with fever, worsening cough, and posttussive emesis. A chest radiograph showed an infiltrate suggestive of bacterial pneumonia in the right lower lobe in association with the cystic lung lesion. Laboratory test results showed an elevated white blood cell count with bandemia. Blood cultures remained negative for bacteria; however, the boy did not defervesce despite the broadening of antibiotic coverage.
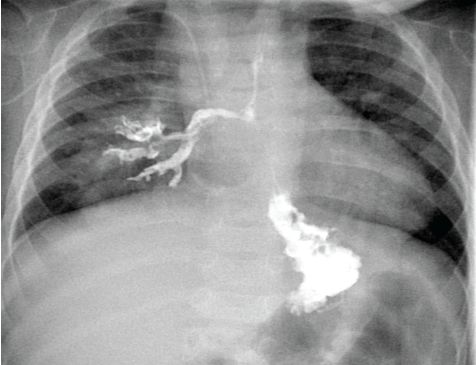
Figure 1. Results of an upper gastrointestinal series in a 7-month-old boy showed contrast material flowing freely from the esophagus into the right lower-lobe bronchus immediately after swallowing.
Given his lack of clinical improvement, he was transferred to our institution for higher level of care.
In consultation with the pediatric surgical service, the recurrent pneumonias involving the right lower lobe were thought to be secondary to the underlying pulmonary malformation, which required resection once the patient was medically stable. His persistent reflux and chronic cough also were worrisome, given that microaspiration may have been contributing to his recurrent pneumonias and lack of clinical improvement. An upper gastrointestinal series was done to further investigate his foregut anatomy. Unexpectedly, the study results demonstrated an esophageal-to-right-lower-lobe-bronchus fistula (Figure 1).
Once he was clinically stable, the patient underwent thoracoscopic resection of the right lower-lobe cystic lesion and repair of the bronchoesophageal fistula. In the operating room, the right lower-lobe lesion was identified, with a fistula communicating with the posterior portion of the mid-esophagus (Figure 2). Also noted was systemic blood supply to the pathologic lung segment, which had not been seen on previous imaging. The intraoperative and pathologic findings were consistent with a communicating bronchopulmonary foregut malformation (CBPFM).
Review of histology results showed tje presence of respiratory epithelium lining the fistula tract. The surrounding lung showed large airways and alveolar spaces, with florid organizing lipoid pneumonia and chronic inflammation, findings consistent with longstanding aspiration of lipid-containing contents from the esophagus into the lung.
The patient’s postoperative course was complicated by a prolonged parenchymal air leak that subsequently resolved with chest-tube management. He recovered well without recurrent infection, reflux, or chronic cough.
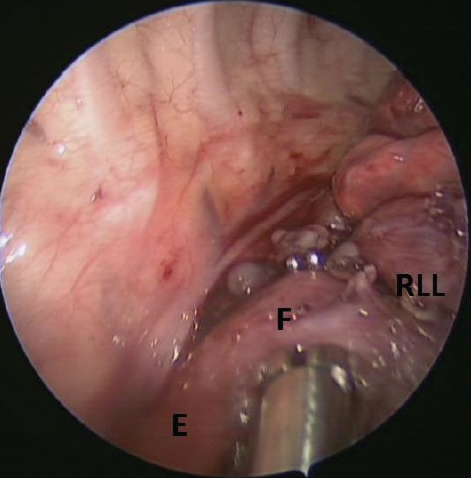
Figure 2. During right thoracoscopy, a fistula (F) was visible between the mid-esophagus (E) and the right lower-lobe bronchus (RLL) before resection.
Discussion
With advances in prenatal care, most developmental malformations of the lung are diagnosed prenatally.1 CPAMs are the most common congenital lung lesions.2 Based on prenatal diagnosis, CPAM has an estimated annual incidence of 1 case per 25,000 to 1 case per 35,000.1
These lesions occur sporadically from unbalanced cellular proliferation and apoptosis during lung development. Postnatally, chest radiography confirms the persistence and extent of the mass, since some will regress prior to delivery.3 The patient then is observed for respiratory distress subsequent to enlargement of the mass from entrapment of air. If symptomatic, urgent resection may be required. Computed tomography often is obtained electively to evaluate the lesion anatomically and to plan operative resection, which may be necessary to prevent infection and malignant degeneration.4
Pulmonary sequestration is a rarer malformation, accounting for 1% to 5% of congenital pulmonary malformations.5 It consists of an area of lung tissue that is nonfunctional, does not communicate with the respiratory tract, and receives its blood supply from systemic rather than pulmonary vasculature. In some cases, this mass of tissue is enveloped in its own visceral layer located outside of the normal lung, known as extralobar sequestration.
Hybrid lesions also can occur in which the sequestered lobe contains tissue with CPAM histology.6 Even more rarely, as seen in this case, the sequestered mass of lung tissue can be connected to the gastrointestinal tract in a CBPFM.7
Four types of CBPFMs have been described: group I, in which a distal CBPFM is associated with esophageal atresia and tracheoesophageal fistula; group II, in which one entire lung originates from the lower esophagus; group III, in which an isolated portion of the lung supplied by pulmonary vasculature communicates with the esophagus or stomach; and group IV, in which the esophagus communicates with a portion of the normal bronchial system that is supplied by systemic rather than pulmonary blood.9
Based on the findings in this case, the patient has a group IV CBPFM, which is the rarest variant, in one review comprising only 5% of the 57 cases reported over 30 years.9
Pathophysiology
The degree and type of CBPFM is influenced by the embryologic stage during which it develops. At approximately 3 weeks’ gestation, the respiratory diverticulum buds from the dorsal foregut. The lateral edges slowly invaginate to form the tracheoesophageal septum, which then pinches off by week 4 to 5 to create separate gastrointestinal and respiratory tubes. A primitive lung bud then emerges from the laryngotracheal tube, which then further develops into 2 bronchopulmonary buds, later forming the right and left lungs. Meanwhile, the gastrointestinal tube develops into the esophagus. Bronchopulmonary foregut malformations arise from a disruption in this sequence of events.
Srikanth and colleagues9 suggest that CBPFM occurs when the lung sacs embrace the lower esophagus and a portion of the lung bud connects to the esophagus through an isolated defect in the mesoderm. This portion then separates from the main group of primordial respiratory cells as it is pulled caudally with the developing esophagus, thus creating a sequestered lobe with a fistula connecting it to the esophagus. Most cases of sequestration do not demonstrate a patent foregut connection, possibly because the sequestration outgrows its blood supply, and the communication involutes.8
Another theory suggests that the respiratory cells that normally derive from the laryngotracheal tube arise directly from the primitive gastrointestinal tube. These cells then separate from the foregut but still may maintain a connection to the esophagus. The cells are unable to maintain normal function as they slowly outgrow their blood supply, which leads to sequestration of the malfunctioning cells.10 The connection itself usually involutes to produce isolated pulmonary sequestration or, as seen in this case, it may persist to produce a CBPFM.
Both of these theories are consistent with the respiratory epithelium lining the fistulous tract seen in this case.
Presentation
Most patients with CBPFM present with respiratory distress that worsens with feeding secondary to aspiration, or present with poor feeding causing failure to thrive.11
Our patient presented with vague symptoms of reflux, chronic cough, and wheezing, which had been attributed to gastroesophageal reflux disease and reactive airway disease exacerbated by his underlying lung pathology. These are very common diagnoses in the pediatric population, especially in children who are growing and developing appropriately, as was the case with our patient.
In the context of his prenatal diagnosis and imaging results, his history of recurrent pneumonia in the area of dysgenic lung tissue also was misleading; the clinical picture was more consistent with a diagnosis of CPAM rather than CBPFM. CPAM also was more likely given the lack of systemic blood supply seen on initial imaging studies. Moreover, chronic infections can cause a sequestered lobe to appear cystic, making it even more difficult to distinguish these entities from each other.
If CBPFM is suspected, the diagnosis can be made with a barium contrast study. As seen in our patient, the contrast enters the esophagus and then flows freely into the bronchi (Figure 1). The management of CBPFM involves surgical resection of the affected pulmonary lobe and repair of the fistula tract.12 The postsurgical prognosis is favorable.13
References:
1.Laberge JM, Flageole H, Pugash D, et al. Outcome of the prenatally diagnosed congenital cystic adenomatoid lung malformation: a Canadian experience. Fetal Diagn Ther. 2001;16(3):178-186.
2.Takeda S, Miyoshi S, Inoue M, et al. Clinical spectrum of congenital cystic disease of the lung in children. Eur J Cardiothorac Surg. 1999; 15(1):11-17.
3.Sauvat F, Michel JL, Benachi A, Emond S, Revillon Y. Management of asymptomatic neonatal cystic adenomatoid malformations. J Pediatr Surg. 2003;38(4):548-552.
4.Priest JR, Williams GM, Hill DA, Dehner LP, Jaffé A. Pulmonary cysts in early childhood and the risk of malignancy. Pediatr Pulmonol. 2009; 44(1):14-30.
5.O’Mara CS, Baker RR, Jeyasingham K. Pulmonary sequestration. Surg Gynecol Obstet. 1978; 147(4):609-616.
6.Liechty KW, Flake AW. Pulmonary vascular malformations. Semin Pediatr Surg. 2008; 17(1):9-16.
7.Clements BS, Warner JO. Pulmonary sequestration and related congenital bronchopulmonary-vascular malformations: nomenclature and classification based on anatomical and embryological considerations. Thorax. 1987;42(6):401-408.
8.Gerle RD, Jaretzki A III, Ashley CA, Berne AS. Congenital bronchopulmonary-foregut malformation — pulmonary sequestration communicating with the gastrointestinal tract. N Engl J Med. 1968;278(26):1413-1419.
9.Srikanth MS, Ford EG, Stanley P, Mahour GH. Communicating bronchopulmonary foregut malformations: classification and embryogenesis. J Pediatr Surg. 1992;27(6):732-736.
10.Leithiser RE Jr, Capitanio MA, Macpherson RI, Wood BP. “Communicating” bronchopulmonary foregut malformations. AJR Am J Roentgenol. 1986;146(2):227-231.
11.Heithoff KB, Sane SM, Williams HJ, et al. Bronchopulmonary foregut malformations: a unifying etiological concept. AJR Am J Roentgenol. 1976;126(1):46-55.
12.Fowler CL, Pokorny WJ, Wagner ML, Kessler MS. Review of bronchopulmonary foregut malformations. J Pediatr Surg. 1988;23(9):793-797.
13.Singal AK, Kumar VR, Rao M, Matthai J. Bilateral communicating bronchopulmonary foregut malformations in a child. Ann Thorac Surg. 2006;82(1):330-332.