
Hypertrophic Cardiomyopathy: Contemporary Evaluation and Management
ABSTRACT: Hypertrophic cardiomyopathy (HCM) is the most common monogenetic hereditary heart disease. With advances in cardiac imaging and genetics, an increasing number of patients with HCM are being identified. However, the condition remains underrecognized, largely because its clinical manifestations are unpredictable and vary among individuals even within the same family. The presentation ranges from sudden cardiac death to a completely benign course with no morbidity or impact on longevity. The overall prognosis of HCM is excellent, but it is this unpredictability that makes the diagnosis and management challenging, requiring a multifaceted approach.
KEYWORDS: Hypertrophic cardiomyopathy, left ventricular outflow tract obstruction, sudden cardiac death
_______________________________________________________________________________________________________________________________________________________
First described in mid-19th century, hypertrophic cardiomyopathy (HCM) is the most common monogenetic hereditary heart disease, affecting 1 in 500 people in the general population.1 Histologic evidence of HCM includes myocyte hypertrophy and fiber disarray with associated interstitial fibrosis. Phenotypic HCM is characterized by asymmetric hypertrophy of the myocardium, more commonly in the interventricular septum, often leading to dynamic obstruction of the left ventricular outflow tract (LVOT).
With several advances in cardiac imaging and genetics, an increasing number of patients with HCM are being identified. However, the condition still remains underrecognized, largely because its clinical manifestations are unpredictable and vary among individuals even within the same family. The presentation ranges from the rare but dreaded sudden cardiac death (SCD) to a completely benign course with no morbidity or impact on longevity. Importantly, the overall prognosis of HCM is excellent, but it is this unpredictability that makes the diagnosis and management challenging, and a multifaceted approach is required.
Clinical evaluation
History and physical examination. Patients with HCM may present with a wide range of symptoms including chest pain, dyspnea, dizziness, fatigue, syncope, or sudden death. It is important to recognize that some patients have minimal or no symptoms. In one HCM cohort study, approximately 70% of the 277 patients studied had mild or no symptoms, and 20% achieved an estimated life expectancy of 70 years or older.2
On physical examination, a systolic murmur may be heard best at the left lower sternal border, which is augmented by maneuvers that decrease preload (Valsalva and squat-to-stand) and is diminished by maneuvers that increase afterload (isometric handgrip and squatting). The presence of cardiomegaly is often noted, the carotid upstroke may be bifid, and an audible S4 may be present.
Electrocardiography (ECG). A 12-lead ECG is recommended during the initial evaluation of all patients with suspected or confirmed HCM. The ECG may show findings of left ventricular hypertrophy (LVH) based on voltage criteria (Sokolov-Lyon criteria are most commonly used—ie, S wave depth in lead V1 or V2 + tallest R wave height in lead V5 or V6 ≥ 35 mm) and widespread Q waves.3,4 These abnormalities, however, do not correlate with severity or pattern of hypertrophy as determined by echocardiography. The exception is with apical variant HCM, where LVH and deep inverted T waves can be seen in leads V2 through V6 as well as in leads II, III, and aVL (Yamaguchi pattern, Figure 1G).5 A normal ECG can be observed in 5% to 10% of patients with HCM; therefore, ECG alone cannot be used to rule out HCM.4
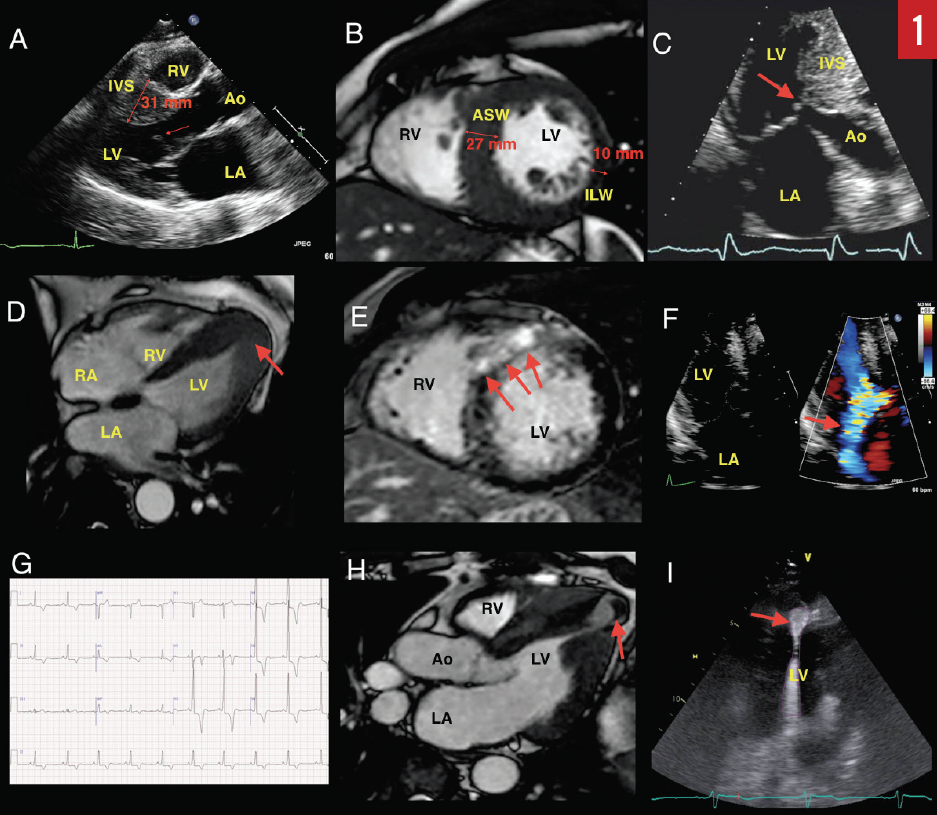
Transthoracic echocardiography (TTE). Echocardiography is the primary modality for evaluating and monitoring patients with suspected or confirmed HCM. Traditionally, morphologic diagnosis is based on the presence of hypertrophy (≥ 15 mm) in the absence of other potential systemic causes for hypertrophy, such as longstanding hypertension or athlete’s heart.3 Milder hypertrophy (> 13 mm) is sometimes considered diagnostic in patients with a positive family history or if other features of HCM are present.6,7 Classically, the basal anteroseptum is involved, but various patterns of hypertrophy involving other areas, as well as diffuse hypertrophy, have also been described.8 A focused view of the apex may identify localized apical hypertrophy (ie, apical variant HCM), which may have associated apical aneurysm (Figure 1D). If present, this can lead to thrombus formation and predispose to thromboembolic events or stroke (Figure 1H).
Often the LV function is hyperdynamic, but rarely patients develop systolic dysfunction—the so-called end-stage or burned-out HCM.9-11 Importantly, increased left atrial size is common and is associated with more frequent cardiovascular adverse events.12 Increased left atrial size correlates with decreased exercise capacity in nonobstructive HCM.13 Approximately one-third of HCM patients have nonobstructive HCM, which appears to have a relatively benign course.14,15
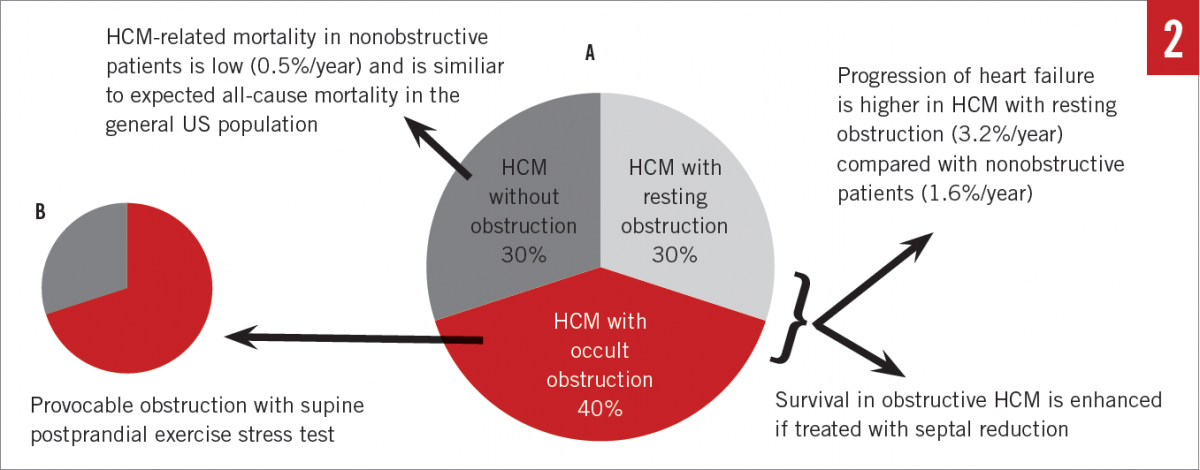
Systolic anterior motion (SAM) of the mitral valve (MV) leaflets is characteristic in hypertrophic obstructive cardiomyopathy (HOCM) and results in characteristic dynamic LVOT obstruction and posteriorly directed mitral regurgitation (MR) (Figures 1A, 1C, and 1F). HCM with significant obstruction at rest (> 30 mm Hg), with provocation, or with exercise (> 50 mm Hg) is present in the majority of patients, with prevalence of up to 70% (Figure 2A).15 Obstruction results from a variety of mechanisms, including narrowing of the outflow from septal hypertrophy, papillary muscle displacement, and elongated MV leaflets.16,17 SAM-septal contact results in a pressure gradient between the LV and the aorta at the subvalvular level and was the very first physiologic abnormality described in the 1950s, resulting in the initial term idiopathic hypertrophic subaortic stenosis.18 Today, it is known that obstruction may occur in the midcavity or apical regions depending on the location of the hypertrophy. In the United States, apical variant HCM is rare and has been noted in 1% to 2% of cases; however, in Japan and other Asian countries, it is more common.8,19,20
Tissue Doppler imaging (TDI). TDI is a useful technique for differentiating HCM from athlete’s heart. Athlete’s heart tends to show normal or super-normal values, whereas there is evidence of abnormal myocardial mechanics in patients with HCM.21 Contemporary evaluation of mechanics using myocardial deformation imaging (strain imaging) suggests that HCM patients have reduced longitudinal but increased circumferential strain.22 LV opacification with contrast echocardiography is important for detecting the presence of apical variant HCM, LV aneurysms, and LV thrombus (Figure 1I). Additionally, contrast echocardiography is used intraprocedurally for the delineation of the perfusion territory of isolated septal perforator arteries prior to alcohol septal ablation (ASA).23 Tissue characterization using myocardial contrast echocardiography is an area of active research.
Stress echocardiography. LVOT obstruction is dynamic in response to a wide range of variables, including volume status, afterload, or preload. While pharmacologic provocation of LVOT obstruction is feasible with administration of amyl nitrite, isoproterenol, or dobutamine, physiologic exercise is always preferable. Exercise stress testing provides a myriad of data regarding functional capacity, occult symptoms, and prognosis.24,25 Moreover, postprandial exercise evaluation has been shown to identify occult LVOT obstruction as a result of splanchnic vasodilation and decreasing afterload in a subset of patients who did not with fasting exercise (Figure 2B).26
Furthermore, evaluation of the hemodynamic response to exercise has implications on risk for SCD and may sway the provider to recommend placement of an implantable cardiac defibrillator (ICD).3 A resting LVOT gradient of greater than 30 mm Hg or provoked LVOT gradient of 50 mm Hg or greater are considered significant, and in the presence of symptoms there is potential to gain improved quality of life as well as longevity through appropriately chosen septal reduction therapies or disopyramide use.27-29
Although not performed routinely in all patients with HCM, metabolic parameters such as anaerobic threshold and peak oxygen consumption can be measured by cardiopulmonary exercise stress testing, which provides comprehensive assessment of functional capacity and can add to the prognostic information in individual patients.25
Transesophageal echocardiography (TEE). TEE is not routinely performed in HCM, but it can be valuable in selected HCM patients with poor acoustic windows on TTE. It can be used for more complete evaluation of the mitral subvalvular apparatus and to help exclude subaortic or supra-aortic membrane. All patients undergoing myectomy benefit from careful surgical planning with multimodality imaging. In many cases, revision of the MV and subvalvular structures is necessary for complete relief of the LVOT obstruction, and intraoperative TEE is a critical tool in the hands of a skilled operator.30
Cardiovascular magnetic resonance imaging (MRI). Cardiovascular MRI has emerged as a vital tool in contemporary management of HCM patients. Stunningly resolute images provide precise localization of hypertrophy, evaluation of the MV apparatus, papillary muscle morphology, and prognostic information from quantification of myocardial fibrosis (Figure 1E).31,32 Cardiovascular MRI has been useful for differentiation of HCM from phenocopies such as hypertensive heart disease, athlete’s heart, and infiltrative and metabolic cardiomyopathies (Figure 1B). Cardiovascular MRI is vital for operative planning and has been used for 3-dimensional printing in order to facilitate this.33
Familial screening and genetic testing. Cascade screening of relatives of patients with HCM is essential. It is recommended that first-degree relatives of the proband undergo serial intermittent ECG and echocardiographic evaluations in the absence of a known disease-causing genetic mutation.3 Phenotypic screening frequency is dependent on the age of the relative, with children aged 7 to 18 years requiring screening every 12 to 18 months, and relatives 18 years and older requiring screening only every 3 to 5 years.
With the advent of commercially available genetic testing, extended and costly phenotypic screening can be avoided in some patients. It can be used to confirm the genetic etiology of a patient’s HCM and to differentiate isolated HCM from other associated syndromic conditions (such as Danon disease, Fabry disease, amyloidosis, and mitochondrial cardiomyopathy).
Most HCM is inherited in an autosomal dominant manner, and first-degree relatives of a person with a known pathogenic HCM gene variant have a 50% risk of having the same disease-causing variant. A significant portion of HCM cases can also be attributed to de novo variants, compound variants, or pathogenic variants in multiple genes; each of these scenarios conveys a different risk to a proband’s relatives.34 Thus, in addition to testing the proband in a family, molecular genetic testing can be used to identify those family members of a proband who are at increased risk for HCM and therefore in need of serial surveillance.
When a definite disease-causing genetic mutation has been identified in a proband, site-specific genotyping of relatives can help identify which individuals require ongoing screening and those who can safely stop. While genotyping is a useful tool for familial risk stratification, there are serious pitfalls that mandate thorough professional genetic counseling and an understanding of the limitations of the technique.
Continue to next page for Management
Management
Treatment is aimed at reducing LVOT obstruction, preventing sudden death, and managing arrhythmias and their risk.
Pharmacotherapy. β-blockers are the usual first-line therapy for symptomatic HOCM.3 For those with mild or no symptoms, β-blockers can mitigate exercise-induced LVOT obstruction.35 While metoprolol is preferred, propranolol and atenolol are acceptable alternatives. Carvedilol and labetalol should be avoided because they can potentially aggravate LVOT obstruction due to α-blocking properties and afterload reduction. Calcium-channel blockers (CCBs) such as diltiazem or verapamil can be used if β-blockers are ineffective or not tolerated as a result of adverse effects. Diltiazem is safe in preclinical HCM, and data exist suggesting that there may be a disease-modifying effect in a subset of genotype-positive HCM patients.36 One should be aware that CCBs can potentially aggravate LVOT obstruction by afterload reduction and should not be used in patients with severe LVOT gradients, and in high doses.37
Disopyramide is a class I anti-arrhythmic with strong negative inotropic effects. As a result, and in combination with either β-blockers or CCBs, disopyramide is the most reliable drug to reduce resting and provocable LVOT obstruction.27 Furthermore, it does not appear to be proarrhythmic in HCM and is an excellent option for refractory symptoms on single-agent therapy.28 When using disopyramide, it is essential to counsel patients on the potential and frequently encountered adverse effects. These include the anticholinergic effects of dry mouth, dry eyes, blurred vision, constipation, and urinary retention (particularly in older men with a history of prostatism).
In addition to considering which are appropriate therapies in HCM, one should also be cognizant of medications that may exacerbate symptoms. Diuretics are usually avoided in HOCM, because the LVOT obstruction can increase in the setting of lowering preload and LV volume. Similarly, vasodilators such as angiotensin-converting enzyme inhibitors and angiotensin receptor blockers, dihydropyridine CCBs, and α-blockers, may increase the LVOT obstruction by decreasing afterload. While recent studies have evaluated the use of angiotensin receptor blockers (losartan and valsartan) in both phenotypic and prephenotypic patients, no significant data have shown proven benefit to date.38,39
Moreover, patients with HOCM may be exposed to serious illness and trauma throughout their lives. It is critical that the treating team in these scenarios is aware of the hemodynamic nuances of HOCM, and that volume resuscitation and α-agonists (phenylephrine) are the backbone of maintaining hemodynamic stability.
Septal reduction and other surgical procedures. Septal reduction therapies should be considered in HCM patients with symptoms attributed to LVOT obstruction. Both ASA and septal myectomy have similar short- and long-term hemodynamic and functional outcomes based on nonrandomized studies but have never been compared head to head. In general, surgical myectomy with appropriate MV revision is the treatment of choice in patients who are able to undergo sternotomy and the associated recovery time. ASA is an important therapy for older patients and those deemed to be at higher risk.3.29 Important limitations of ASA include the technical feasibility based on anatomic limitations, the inability to address abnormalities related to the MV, and an overall higher risk for development of complete heart block.40,41
MV and related apparatus abnormalities are common in HCM, leading to MR and worsening LVOT obstruction with SAM.42,43 MV repair and subvalvular revision is preferred over replacement. Techniques aimed at addressing abnormalities in the MV and subvalvular revision include anterior MV plication, papillary muscle realignment, chordal transposition, and synthetic chord placement; very occasionally, annuloplasty rings are used if the patient has intrinsic MV disease.
More recently, several patients with SAM-septal contact and symptomatic LVOT obstruction have been successfully treated with percutaneous MV repair using a transcatheter MV clip device (MitraClip, Abbott).44-47 Our center and others have reported encouraging results.48
Only in rare instances is MV replacement required, and our experience is that mechanical valves are a more appropriate choice in such cases. Subvalvular protrusion of the bioprosthetic valve may result in a neo-LVOT and a fixed LVOT obstruction, particularly in patients with small LV cavities.
In selected cases where medical therapies fail, and septal reduction is contraindicated, preferential right ventricular pacing has the potential to reduce LVOT obstruction and relieve symptoms.49-51 This strategy should not be routinely used, since chronic and persistent right ventricular pacing has also been associated with reduced LV ejection fraction.52
SCD prediction and risk mitigation
Sudden death is the most dreaded manifestation of HCM. While overall it is rare in HCM (< 1%/year), it occurs more frequently in younger patients. SCD is attributable to ventricular arrhythmias and thus is preventable with the appropriate use of ICDs. Subcutaneous ICDs are also a consideration and can reduce the burden on intravascular hardware in select patients.53 No evidence supports pharmacotherapy, including antiarrhythmics, to prevent SCD. The use of antiarrhythmics in nonsustained ventricular tachycardia is controversial.
Identifying patients with a higher SCD risk is one of the greatest challenges, since ICD implantation, especially in young patients, is by no means without consequence. Importantly, restratification for SCD risk is necessary on an annual basis. Traditional risk factors have been well documented and include the following3,54:
- A personal history of sudden cardiac arrest or ventricular tachyarrhythmia (ventricular fibrillation, sustained ventricular tachycardia)
- Nonsustained ventricular tachycardia (> 3 beats at > 120 beats/min on a Holter monitor)
- Massive hypertrophy (≥ 30 mm)
- Recent unexplained syncope
- A first-degree relative with sudden unexplained death
- A hypotensive/flat hemodynamic response to exercise
Despite these being well proven and lifesaving markers, many patients undergo ICD placement unnecessarily. Refinement of SCD risk assessment has been a subject of intense research that has resulted in several newly evolving markers. Among these are quantification of fibrosis on cardiovascular MRI55-57; LVOT gradient of 30 mm Hg or more54,58,59; apical aneurysm19,60; double or compound pathogenic sarcomeric gene variants34; and impaired coronary flow (myocardial ischemia).61,62
Recently, O’Mahony and colleagues have published a novel risk calculator that utilizes a number of established risk markers as well as a patient’s age to generate a numeric risk.63 While this method has not been prospectively studied, it is endorsed by the European Society of Cardiology and can be a helpful tool when discussing risk with patients and family members.64 A shared decision model between the patient and the health care provider, taking into account the individual philosophy and risk tolerance, is essential.
Atrial arrhythmias. Atrial fibrillation and atrial flutter (AF) are common in HCM, particularly as patients age. It is often symptomatic and poorly tolerated. Stroke risk is much higher compared with standard nonvalvular AF, and as such thromboembolic risk calculators (eg, CHADS2, CHA2DS2VASc) are not applicable in HCM.65-67 In general, systemic anticoagulation is recommended unless it is specifically contraindicated. Given the hemodynamic effects of loss of atrial contraction, a rhythm-control strategy is preferred. Electrical cardioversion, antiarrhythmic medications, pulmonary vein isolation, and surgical maze and left atrial appendage ligation at the time of surgical myectomy are useful treatment options.
When considering antiarrhythmic drugs, disopyramide is a good option in patients with concomitant LVOT obstruction. Of note, these patients need to be on an adequate dose of an atrioventricular (AV)-nodal blocking agent in addition to disopyramide in order to avoid paradoxical shortened conduction and rapid ventricular rates. Amiodarone is also effective, but the dose-dependent likelihood of drug toxicity is a concern in younger patients. Rate control with β-blockers or CCBs can be tried if rhythm control fails. Importantly, digoxin should be avoided due to the positive inotropic effect and risk for worsening LVOT obstruction. Rarely, in refractory symptomatic AF, AV-nodal ablation and pacemaker placement can be employed.
Exercise and lifestyle in HCM. The prevailing opinion of many HCM experts is that regular moderate-intensity exercise is recommended for individuals with HCM. While there are no prospective data to support this, outcomes from an ongoing study evaluating exercise patterns in HCM (Lifestyle and Exercise in Hypertrophic Cardiomyopathy trial, or LIVE-HCM) are eagerly awaited. Participation in high-intensity and competitive-level activities (particularly burst activities) is strongly discouraged.3 Obesity with concomitant HCM can present as a downward spiral of worsening exertional intolerance and subsequent increasing body mass. It is important to address this early and repeatedly. Obstructive sleep apnea is common in HCM (31%-71%) and is associated with worse outcomes.68-70 There is a lack of data to show that treatment with continuous-positive airway pressure is effective, and further study is indicated. Caffeine (positive inotropy), alcohol, (peripheral vasodilation), and rich foods (splanchnic vasodilation) can worsen LVOT obstruction and should be avoided when possible. Instructions on maintaining hydration, particularly with common illnesses, may help patients avoid hospital admission.
Psychosocial challenges in HCM. Patients with genetically mediated HCM face the chronic, progressive, hereditary nature of their disease, living with an ICD, undergoing invasive procedures, and facing the specter of sudden death.71 Adults relay significant concern about transmission of genetic disease risk to their descendants.72 Not surprisingly, many patients with HCM report anxiety, depression, and posttraumatic stress related to ICDs.73 On the other hand, surgical relief of obstructive symptoms is associated with a decrease in depression and anxiety.74-76 The impact of exercise and sports restriction can be emotionally devastating in HCM patients.77,78 An effective means of ameliorating some of these issues is the incorporation of a genetic counselor into the consultative machinery of the clinic.79
The Take-Home Message
HCM is a phenotypically heterogeneous cardiac condition with a multitude of physiologic effects, most of which have an effective contemporary therapy available, making this an exciting and rewarding condition to treat in medicine. Patients with HCM now enjoy longevity and are subsequently exposed not only to the risks and morbidity of their disease, but also to the risks and morbidity of the self-same therapies and procedures responsible for their longevity. Moreover, it is important to maintain humanity and compassion with our patients. Their condition is chronic, progressive, lifestyle changing, and involves many of the people closest to them—often resulting in psychology overshadowing the physiology.
Suwen Kumar, MD, is a fellow in advanced cardiac imaging at the Knight Cardiovascular Institute at Oregon Health and Science University (OHSU) in Portland, Oregon.
Howard Song, MD, PhD, is chief of the Division of Cardiothoracic Surgery at the Knight Cardiovascular Institute at OHSU in Portland, Oregon.
Meghan Chirpich, MS, CGC, is a genetic counselor at the Knight Cardiovascular Institute at OHSU in Portland, Oregon.
Stephen Heitner, MD, is a cardiology specialist at the Knight Cardiovascular Institute at OHSU in Portland, Oregon.
References:
- Maron BJ, Ommen SR, Semsarian C, Spirito P, Olivotto I, Maron MS. Hypertrophic cardiomyopathy: present and future, with translation into contemporary cardiovascular medicine. J Am Coll Cardiol. 2014;64(1):83-99.
- Maron BJ, Casey SA, Poliac LC, Gohman TE, Almquist AK, Aeppli DM. Clinical course of hypertrophic cardiomyopathy in a regional United States cohort. JAMA. 1999;281(7):650-655.
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2011;58(25):e212-e260.
- McLeod CJ, Ackerman MJ, Nishimura RA, Tajik AJ, Gersh BJ, Ommen SR. Outcome of patients with hypertrophic cardiomyopathy and a normal electrocardiogram. J Am Coll Cardiol. 2009;54(3):229-233.
- Wigle ED. The diagnosis of hypertrophic cardiomyopathy. Heart. 2001;86(6):709-714.
- Maron MS, Maron BJ, Harrigan C, et al. Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J Am Coll Cardiol. 2009;54(3):220-228.
- Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013;381(9862):242-255.
- Klues HG, Schiffers A, Maron BJ. Phenotypic spectrum and patterns of left ventricular hypertrophy in hypertrophic cardiomyopathy: morphologic observations and significance as assessed by two-dimensional echocardiography in 600 patients. J Am Coll Cardiol. 1995;26(7):1699-1708.
- Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation. 2006;114(3):216-225.
- Cecchi F, Sgalambro A, Baldi M, et al. Microvascular dysfunction, myocardial ischemia, and progression to heart failure in patients with hypertrophic cardiomyopathy. J Cardiovasc Transl Res. 2009;2(4):452-461.
- Fujiwara H, Onodera T, Tanaka M, et al. Progression from hypertrophic obstructive cardiomyopathy to typical dilated cardiomyopathy-like features in the end stage. Jpn Circ J. 1984;48(11):1210-1214.
- Yang H, Woo A, Monakier D, et al. Enlarged left atrial volume in hypertrophic cardiomyopathy: a marker for disease severity. J Am Soc Echocardiogr. 2005;18(10):1074-1082.
- Sachdev V, Shizukuda Y, Brenneman CL, et al. Left atrial volumetric remodeling is predictive of functional capacity in nonobstructive hypertrophic cardiomyopathy. Am Heart J. 2005;149(4):730-736.
- Maron MS, Rowin EJ, Olivotto I, et al. Contemporary natural history and management of nonobstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2016;67(12):1399-1409.
- Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation. 2006;114(21):2232-2239.
- He S, Hopmeyer J, Lefebvre XP, Schwammenthal E, Yoganathan AP, Levine RA. Importance of leaflet elongation in causing systolic anterior motion of the mitral valve. J Heart Valve Dis. 1997;6(2):149-159.
- Izgi C, Akgun T, Men EE, Feray H. Systolic anterior motion of the mitral valve in the absence left ventricular hypertrophy: role of mitral leaflet elongation and papillary muscle displacement. Echocardiography. 2010;27(4):E36-E38.
- Braunwald E, Lambrew CT, Rockoff SD, Ross J Jr, Morrow AG. Idiopathic hypertrophic subaortic stenosis: I. A description of the disease based upon an analysis of 64 patients. Circulation. 1964;30(5 suppl 4):3-119.
- Maron MS, Finley JJ, Bos JM, et al. Prevalence, clinical significance, and natural history of left ventricular apical aneurysms in hypertrophic cardiomyopathy. Circulation. 2008;118(15):1541-1549.
- Kitaoka H, Doi Y, Casey SA, Hitomi N, Furuno T, Maron BJ. Comparison of prevalence of apical hypertrophic cardiomyopathy in Japan and the United States. Am J Cardiol. 2003;92(10):1183-1186.
- Vinereanu D, Florescu N, Sculthorpe N, Tweddel AC, Stephens MR, Fraser AG. Differentiation between pathologic and physiologic left ventricular hypertrophy by tissue Doppler assessment of long-axis function in patients with hypertrophic cardiomyopathy or systemic hypertension and in athletes. Am J Cardiol. 2001;88(1):53-58.
- Yang H, Sun JP, Lever HM, et al. Use of strain imaging in detecting segmental dysfunction in patients with hypertrophic cardiomyopathy. J Am Soc Echocardiogr. 2003;16(3):233-239.
- Alam M, Dokainish H, Lakkis N. Alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a systematic review of published studies. J Interv Cardiol. 2006;19(4):319-327.
- Desai MY, Bhonsale A, Patel P, et al. Exercise echocardiography in asymptomatic HCM: exercise capacity, and not LV outflow tract gradient predicts long-term outcomes. JACC Cardiovasc Imaging. 2014;7(1):26-36.
- Masri A, Pierson LM, Smedira NG, et al. Predictors of long-term outcomes in patients with hypertrophic cardiomyopathy undergoing cardiopulmonary stress testing and echocardiography. Am Heart J. 2015;169(5):684-692.e1.
- Feiner E, Arabadjian M, Winson G, Kim B, Chaudhry F, Sherrid MV. Post-prandial upright exercise echocardiography in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2013;61(24):2487-2488.
- Verlinden NJ, Coons JC. Disopyramide for hypertrophic cardiomyopathy: a pragmatic reappraisal of an old drug. Pharmacotherapy. 2015;35(12):1164-1172.
- Sherrid MV, Barac I, McKenna WJ, et al. Multicenter study of the efficacy and safety of disopyramide in obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2005;45(8):1251-1258.
- Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2005;46(3):470-476.
- Sherrid MV, Balaram S, Kim B, Axel L, Swistel DG. The mitral valve in obstructive hypertrophic cardiomyopathy: a test in context. J Am Coll Cardiol. 2016;67(15):1846-1858.
- Chan RH, Maron BJ, Olivotto I, et al. Prognostic value of quantitative contrast-enhanced cardiovascular magnetic resonance for the evaluation of sudden death risk in patients with hypertrophic cardiomyopathy. Circulation. 2014;130(6):484-495.
- Maron MS, Maron BJ. Clinical impact of contemporary cardiovascular magnetic resonance imaging in hypertrophic cardiomyopathy. Circulation. 2015;132(4):292-298.
- Yang DH, Kang J-W, Kim N, Song J-K, Lee J-W, Lim T-H. Myocardial 3-dimensional printing for septal myectomy guidance in a patient with obstructive hypertrophic cardiomyopathy. Circulation. 2015;132(4):300-301.
- Maron BJ, Maron MS, Semsarian C. Double or compound sarcomere mutations in hypertrophic cardiomyopathy: a potential link to sudden death in the absence of conventional risk factors. Heart Rhythm. 2012;9(1):57-63.
- Nistri S, Olivotto I, Maron MS, et al. β blockers for prevention of exercise-induced left ventricular outflow tract obstruction in patients with hypertrophic cardiomyopathy. Am J Cardiol. 2012;110(5):715-719.
- Ho CY, Lakdawala NK, Cirino AL, et al. Diltiazem treatment for pre-clinical hypertrophic cardiomyopathy sarcomere mutation carriers: a pilot randomized trial to modify disease expression. JACC Heart Fail. 2015;3(2):180-188.
- Epstein SE, Rosing DR. Verapamil: its potential for causing serious complications in patients with hypertrophic cardiomyopathy. Circulation. 1981;64(3):437-441.
- Olivotto I, Ashley EA. INHERIT (INHibition of the renin angiotensin system in hypertrophic cardiomyopathy and the Effect on hypertrophy—a Randomised Intervention Trial with losartan). Glob Cardiol Sci Pract. 2015;2015:7. doi:10.5339/gcsp.2015.7
- Shimada YJ, Passeri JJ, Baggish AL, et al. Effects of losartan on left ventricular hypertrophy and fibrosis in patients with nonobstructive hypertrophic cardiomyopathy. JACC Heart Fail. 2013;1(6):480-487.
- Leonardi RA, Kransdorf EP, Simel DL, Wang A. Meta-analyses of septal reduction therapies for obstructive hypertrophic cardiomyopathy: comparative rates of overall mortality and sudden cardiac death after treatment. Circ Cardiovasc Interv. 2010;3(2):97-104.
- Liebregts M, Vriesendorp PA, Mahmoodi BK, Schinkel AFL, Michels M, ten Berg JM. A systematic review and meta-analysis of long-term outcomes after septal reduction therapy in patients with hypertrophic cardiomyopathy. JACC Heart Fail. 2015;3(11):896-905.
- Kwon DH, Setser RM, Thamilarasan M, et al. Abnormal papillary muscle morphology is independently associated with increased left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. Heart. 2008;94(10):1295-1301.
- Harrigan CJ, Appelbaum E, Maron BJ, et al. Significance of papillary muscle abnormalities identified by cardiovascular magnetic resonance in hypertrophic cardiomyopathy. Am J Cardiol. 2008;101(5):668-673.
- Sorajja P, Pedersen WA, Bae R, et al. First experience with percutaneous mitral valve plication as primary therapy for symptomatic obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2016;67(24):2811-2818.
- Pantazis A, Cheang MH, Mullen M, et al. 94 percutaneous mitral repair in hypertrophic cardiomyopathy [abstract]. Heart. 2014;100(suppl 3):A54-A55.
- Schäfer U, Frerker C, Thielsen T, et al. Targeting systolic anterior motion and left ventricular outflow tract obstruction in hypertrophic obstructed cardiomyopathy with a MitraClip. EuroIntervention. 2015;11(8):942-947.
- Schäfer U, Kreidel F, Frerker C. MitraClip implantation as a new treatment strategy against systolic anterior motion-induced outflow tract obstruction in hypertrophic obstructive cardiomyopathy. Heart Lung Circ. 2014;23(5):e131-e135.
- Gupta S, Slater M, Wei K, Heitner S. Percutaneous mitral valve repair for high-risk symptomatic hypertrophic obstructive cardiomyopathy. Presented at: Cardiovascular Research Foundation 9th Annual Transcatheter Valve Therapies Conference; June 16-18, 2016; Chicago, IL.
- Gadler F, Linde C, Juhlin-Dannfeldt A, Ribeiro A, Rydén L. Influence of right ventricular pacing site on left ventricular outflow tract obstruction in patients with hypertrophic obstructive cardiomyopathy. J Am Coll Cardiol. 1996;27(5):1219-1224.
- Fifer MA, Vlahakes GJ. Management of symptoms in hypertrophic cardiomyopathy. Circulation. 2008;117(3):429-439.
- Kepski R, Buchner T, Cytowski J, Malecka L, Walczak F. Adaptive filtering in exercise high resolution ECG as applied to the hypertrophic cardiomyopathy. Pacing Clin Electrophysiol. 2001;24(8 pt 1):1216-1223.
- Tops LF, Schalij MJ, Bax JJ. The effects of right ventricular apical pacing on ventricular function and dyssynchrony implications for therapy. J Am Coll Cardiol. 2009;54(9):764-776.
- Weinstock J, Bader YH, Maron MS, Rowin EJ, Link MS. Subcutaneous implantable cardioverter defibrillator in patients with hypertrophic cardiomyopathy: an initial experience. J Am Heart Assoc. 2016;5(2):e002488.
- Maki S, Ikeda H, Muro A, et al. Predictors of sudden cardiac death in hypertrophic cardiomyopathy. Am J Cardiol. 1998;82(6):774-778.
- Rubinshtein R, Glockner JF, Ommen SR, et al. Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail. 2010;3(1):51-58.
- Moon JCC, McKenna WJ, McCrohon JA, Elliott PM, Smith GC, Pennell DJ. Toward clinical risk assessment in hypertrophic cardiomyopathy with gadolinium cardiovascular magnetic resonance. J Am Coll Cardiol. 2003;41(9):1561-1567.
- Adabag AS, Maron BJ, Appelbaum E, et al. Occurrence and frequency of arrhythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. J Am Coll Cardiol. 2008;51(14):1369-1374.
- Maron MS, Olivotto I, Betocchi S, et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med. 2003;348(4):295-303.
- Elliott PM, Gimeno JR, Tomé MT, et al. Left ventricular outflow tract obstruction and sudden death risk in patients with hypertrophic cardiomyopathy. Eur Heart J. 2006;27(16):1933-1941.
- Paul M, Schäfers M, Grude M, et al. Idiopathic left ventricular aneurysm and sudden cardiac death in young adults. Europace. 2006;8(8):607-612.
- Cecchi F, Olivotto I, Gistri R, Lorenzoni R, Chiriatti G, Camici PG. Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. N Engl J Med. 2003;349(11):1027-1035.
- Sorajja P, Ommen SR, Nishimura RA, Gersh BJ, Berger PB, Tajik AJ. Adverse prognosis of patients with hypertrophic cardiomyopathy who have epicardial coronary artery disease. Circulation. 2003;108(19):2342-2348.
- O’Mahony C, Jichi F, Pavlou M, et al; Hypertrophic Cardiomyopathy Outcomes Investigators. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J. 2014;35(30):2010-2020.
- Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(39):2733-2779.
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation. 2001;104(21):2517-2524.
- Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart. 2014;100(6):465-472.
- Guttmann OP, Pavlou M, O’Mahony C, et al; Hypertrophic Cardiomyopathy Outcomes Investigators. Prediction of thrombo-embolic risk in patients with hypertrophic cardiomyopathy (HCM Risk-CVA). Eur J Heart Fail. 2015;17(8):837-845.
- Banno K, Shiomi T, Sasanabe R, et al. Sleep-disordered breathing in patients with idiopathic cardiomyopathy. Circ J. 2004;68(4):338-342.
- Pedrosa RP, Drager LF, Genta PR, et al. Obstructive sleep apnea is common and independently associated with atrial fibrillation in patients with hypertrophic cardiomyopathy. Chest. 2010;137(5):1078-1084.
- Konecny T, Somers VK. Sleep-disordered breathing in hypertrophic cardiomyopathy: challenges and opportunities. Chest. 2014;146(1):228-234.
- Caleshu C, Kasparian NA, Edwards KS, et al. Interdisciplinary psychosocial care for families with inherited cardiovascular diseases. Trends Cardiovasc Med. 2016;26(7):647-653.
- Aatre RD, Day SM. Psychological issues in genetic testing for inherited cardiovascular diseases. Circ Cardiovasc Genet. 2011;4(1):81-90.
- Ingles J, Sarina T, Kasparian N, Semsarian C. Psychological wellbeing and posttraumatic stress associated with implantable cardioverter defibrillator therapy in young adults with genetic heart disease. Int J Cardiol. 2013;168(4):3779-3784.
- Bensch J, Huebner M, Minnier J, Slater M, Cigarroa J, Heitner S. Septal myectomy improves symptoms more than alcohol septal ablation in patients with hypertrophic obstructive cardiomyopathy [abstract]. J Am Coll Cardiol. 2016;67(13 suppl):1523.
- Serber ER, Sears SF, Nielsen CD, Spencer WH III, Smith KM. Depression, anxiety, and quality of life in patients with obstructive hypertrophic cardiomyopathy three months after alcohol septal ablation. Am J Cardiol. 2007;100(10):1592-1597.
- ten Cate FJ, Soliman OII, Michels M, et al. Long-term outcome of alcohol septal ablation in patients with obstructive hypertrophic cardiomyopathy: a word of caution. Circ Heart Fail. 2010;3(3):362-369.
- Reineck E, Rolston B, Bragg-Gresham JL, et al. Physical activity and other health behaviors in adults with hypertrophic cardiomyopathy. Am J Cardiol. 2013;111(7):1034-1039.
- Asif IM, Price DE, Ewing A, Rao AL, Harmon KG, Drezner JA. The impact of diagnosis: measuring the psychological response to being diagnosed with serious or potentially lethal cardiac disease in young competitive athletes. Br J Sports Med. 2016;50(3):163-166.
- Ingles J, Lind JM, Phongsavan P, Semsarian C. Psychosocial impact of specialized cardiac genetic clinics for hypertrophic cardiomyopathy. Genet Med. 2008;10(2):117-120.