
A Collection of Obscure Syndromes
Purple Urine Bag Syndrome
Edwin Pimentel-Brugal, MD, Moise Carrington, MD, Laura Andrews, RPh, CGP, and Hampton Groover, MD
An 85-year-old woman who was receiving hospice care noticed purple discoloration in the tubing and urine bag of her urinary catheter. The patient had metastatic breast cancer associated with malnutrition, dehydration, recurrent urinary tract infection (UTI), and chronic opiate-related constipation and nausea.
Laboratory testing. Urinalysis revealed ketones, positive nitrite, 1 to 3 white blood cells per high-power field, 4+ bacteria, and normal pH; urine culture grew more than 100,000 colony-forming units/mL of Escherichia coli and Morganella morganii. The patient was treated for the infection.

Discussion. Purple urine bag syndrome is associated with UTI in catheterized patients, usually elderly women with significant comorbidities.1,2 Although the syndrome is rare, a prevalence as high as 9.8% has been reported among patients in long-term care facilities.2,3 Alkaline urine and dehydration are common cofactors.2
Like most frail elderly persons with UTI, affected patients are often asymptomatic.4 Gram-negative bacterial strains that possess sulphatase or phosphatase cause the conversion of indican, a tryptophan metabolite, into indirubin (red) and indigo (blue), which produce the urinary pigments.1,2 The catheter drainage system is progressively stained from red to violet by the pigments, while the urine itself is not grossly discolored. The rarity of purple urine bag syndrome, despite the high prevalence of UTI, may signify a lack of these enzymes in other bacteria of the same species.
Discoloration of urine collection systems is uncommon and may indicate significant underlying pathology. Awareness of this unique presentation can help identify gram-negative UTI in a patient population with otherwise classically vague, delayed, or absent symptoms.
References:
1. Ribeiro JP, Marcelino P, Marum S, et al. Case report: purple urine bag syndrome. Crit Care. 2004;8(3):R137.
2. Harun NS, Nainar SK, Chong VH. Purple urine bag syndrome: a rare and interesting phenomenon. South Med J. 2007;100(10):1048-1050.
3. Schneiderman H. Purple urine bags syndrome: another complication of chronic urethral catheterization. Consultant. 2006;46:787-792.
4. Su FH, Chung SY, Chen MH, et al. Case analysis of purple urine-bag syndrome at a long-term care service in a community hospital. Chang Gung Med J. 2005;28(9):636-642.
Bilateral Alien Hand Syndrome
Kathleen Sheridan, DO, Brian Johnson, MD, Zoe Maher, MD, and Kofi Clarke, MD
A 68-year-old woman was hospitalized because of confusion and agitation of sudden onset. Her history included dementia and multiple infarcts of both cerebellar hemispheres, bilateral basal ganglia, bilateral parietal lobes, and the right occipital lobe.
In the emergency department, the patient made erratic grasping movements with her right (dominant) hand and stated that her arm was “being pulled by a ghost.” She said that she was unable to control her left arm and was concerned that she would harm herself or others. For example, she attempted to choke one of the examiners with the left hand, and she wrapped a call button cord around her neck with the same hand. She also had left lower extremity weakness of recent onset.
Physical examination. The patient was oriented to person and place but could name only 3 of 5 objects held in front of her. Asymmetry of the left nasolabial fold was noted. Sensory and visual field findings were unremarkable. There was mild to moderate left hemiparesis. Reflexes were symmetrical with equivocal plantar responses. She was unable to name any of 5 objects placed in her left hand but correctly identified all 5 objects placed in her right hand. She could not distinguish her left hand from the examiner’s hand or point to her own body parts using the left hand, but she was able to do so with the right hand.
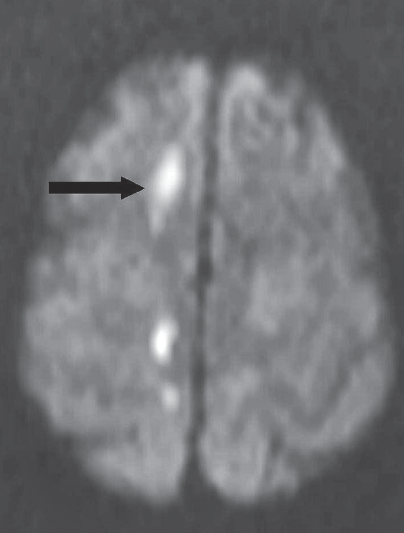
Laboratory tests. MRI scans of the head revealed acute infarcts (arrows) in the right corona radiata, right centrum semiovale, and anterior and posterior corpus callosum.
Discussion. The diagnosis based on neurological consultation was alien hand syndrome, which is the loss of control of 1 or both upper extremities when the involuntary movements of the affected extremity are not caused by a movement disorder.1 Several anatomical lesions have been identified as causes: the corpus callosum alone, the corpus callosum plus the dominant medial frontal cortex, and the posterior cortical and subcortical regions. The posterior form of the syndrome typically results from corticobasal degeneration or from a cerebrovascular accident.2 The anterior forms are associated with anterior cerebral artery territory infarction.
Lesions that involve the corpus callosum alone typically present with the hands acting in conflict with each other (intermanual conflict): for example, one hand would attempt to button a shirt, while the other hand would undo the buttons. Limb paresis is typically not present.2
Frontocallosal injury results in compulsive grasping behaviors. Generally, the dominant ipsilateral hand is affected. It is hypothesized that transcortical inhibition from the right frontal lobe onto the left supplementary motor area must be impaired for the manifestation of a right dominant alien hand.3 There is associated paresis of the contralateral leg as well.2
The posterior form of alien hand syndrome typically involves the nondominant hand. This hand displays levitation and nonpurposeful movements. There is a tendency to assume abnormal postures when corticobasal degeneration is present. Sensory impairment and body schema distortion are found in patients with this lesion.2
Outcome of the case. In this patient, the syndrome manifested by her left hand can be attributed to the posterior callosal infarct, while the grasping nature of her dominant right “alien” hand may result from a new anterior callosal infarct combined with the prior parietal lobe infarct. Despite the association of this syndrome with Alzheimer-type dementia, we believe our patient’s condition was related to the new infarcts. ■
References:
1. Geschwind DH, Iacoboni M, Mega MS, et al. Alien hand syndrome: interhemispheric motor disconnection due to a lesion in the midbody of the corpus callosum. Neurology. 1995;45(4):802-808.
2. Marey-Lopez J, Rubio-Nazabal E, Alonso-Magdalena L, Lopez-Facal S. Posterior alien hand syndrome after a right thalamic infarct.
J Neurol Neurosurg Psychiatry. 2002;73(4):447-449.
3. Feinberg TE, Schindler RJ, Flanagan NG, Haber LD. Two alien hand syndromes. Neurology. 1992;42(1):19-24.
Horner Syndrome
D. Brady Pregerson, MD
A 47-year-old man presented to the emergency department with a drooping right eye. He also complained of a constant right-sided headache of 1 week’s duration. The pain involved the temporal region. Another physician had diagnosed new-onset migraine and prescribed sumatriptan, which failed to alleviate the pain.
Physical examination. The patient had no weakness, vomiting, or double vision. Both his father and his son had Marfan syndrome. The patient’s vital signs were normal. Mild right-sided ptosis and miosis—accentuated by dim light—were noted. Cranial nerves were otherwise intact, and no other neurologic abnormalities were found. The neck was supple without bruits. Breath sounds were normal. There was no digital clubbing.

Diagnostic studies. A chest radiograph and noncontrast CT scan of the brain were normal. Carotid dissection and subarachnoid hemorrhage were considered in the differential. Analysis of cerebrospinal fluid from a lumbar puncture showed no white blood cells, 3 to 6 red blood cells per microliter, a glucose level of 55 mg/dL, and a protein level of 53 mg/dL. These findings ruled out subarachnoid hemorrhage. During the patient’s hospital stay, a CT scan of the chest was negative for Pancoast tumor of the lung.
An MRI scan/magnetic resonance angiogram of the brain showed caliber irregularity and slight course derangement of the high cervical, petrous portion of the right internal carotid artery. Long-segment atherosclerosis was considered unlikely because there was no other site of atherosclerotic change. Fibromuscular dysplasia was also considered. Carotid dissection was strongly suspected. An angiogram of the brain performed a month later showed a 2.5 cm long segment of mild irregularity and focal ulceration in the middle right internal carotid artery, with no definite evidence of a focal arterial dissection in this area.
Discussion. Horner syndrome—a functional sympathectomy of the ipsilateral eye—is caused by injury or disruption of the neural plexus that runs from the sympathetic chain, past the apex of the lung, and up the carotid artery to the eye. Ptosis may be subtle. Miosis is more marked in dim light; it may be difficult to notice in bright light. Potential causes of Horner syndrome include carotid or vertebral
artery dissection, aortic dissection, traumatic carotid injury, deep neck infections, cerebrovascular accident, cerebellar bleed, cluster headache, and Pancoast tumor of the lung.
Initial symptoms of carotid dissection usually involve pain that affects one side of the neck, face, or head; the pain may start abruptly but usually the onset is gradual. Pulsatile tinnitus or a bruit is present in about 25% of affected patients. Early neurologic findings may involve the sympathetic plexus (Horner syndrome is present in about 50% of patients), cranial nerve XII, or cerebellar function. Eventually, transient ischemic attack or thrombotic stroke may occur. The mean time between onset of pain and onset of neurologic symptoms is 4 days. Carotid dissection is an important cause of stroke in young adults and accounts for up to 25% of cases.1 If carotid dissection is suspected, magnetic resonance angiography (MRA) with fat suppression is the “gold standard” test. If MRA is not available, alternative tests include carotid duplex ultrasonography and CT angiography (which is almost as sensitive as MRA).
Treatment. Treatment of suspected carotid dissection involves consultation with a neurologist and a neurosurgeon. In contrast to aortic dissection, thrombosis—not rupture—causes complications, and treatment with standard-dose heparin is started in suspicious cases, following a negative head CT scan. Anticoagulation is usually maintained for 3 months. Surgery is rarely required.
Outcome of the case. This patient almost certainly had a carotid dissection related to subclinical Marfan syndrome. Although his imaging studies were nondiagnostic, they were highly suggestive of these disorders. He probably should have received anticoagulant therapy for 3 months. Nevertheless, he did well and did not have a stroke.
His Horner syndrome resolved after 3 months, although he complained of occasional mild right temporal headaches.
Reference:
1. Chaves C, Estol C, Esnaola MM, et al. Spontaneous intracranial internal carotid artery dissection:report of 10 patients. Arch Neurol. 2002;59(6):977-981.
Waardenburg Syndrome
Les Trope, MD, Hillel Trope, MD, and Shlomo Trope, MD
During a routine physical examination, a white forelock was noted on a 54-year-old man. The patient stated that the discolored patch of hair had been present since adolescence. Other than mild hearing loss, he had no significant personal or family medical history. The physicians diagnosed the patient with Waardenburg syndrome.

Discussion. This autosomal dominant condition is usually associated with the following: pigmentary variations (such as white eyelashes), a frontal, white lock of hair, heterochromia iridis, wide bridge of the nose, and cochlear deafness.
The diagnosis in this case was based on clinical appearance. The patient had mild sensorineural deafness; however, his eyes did not show any pigmentary differences. Petrus Johannes Waardenburg of Holland described this syndrome in 1954. He found that the syndrome affects about 1.4% of congenitally deaf children (an overall incidence of 1 in 42,000).
There are at least 4 types of Waardenburg syndrome; the type is determined based on the patient’s physical characteristics. Rarely, Waardenburg syndrome has been associated with intestinal disorders, elevation of the shoulder blade, and spinal disorders. In these cases, genetic counseling should be considered. Four different genes for Waardenburg syndrome have been identified: PAX3, MITF, EDNRB, and EDN3. PAX3, the most common, is located on chromosome 2. Because this is a dominant gene, there is a 50% chance that a child of a person with this syndrome will also have this syndrome.
Treatment. Although no treatment is available for Waardenburg syndrome, recognition of this condition is important.
“Friendly Dog” Syndrome
William B. Wadlington, MD
The wounds on the back of this boy’s head resulted from an encounter with his neighbor’s dog. The youngster’s anxious parents brought him in for evaluation 1 hour after he was bitten.
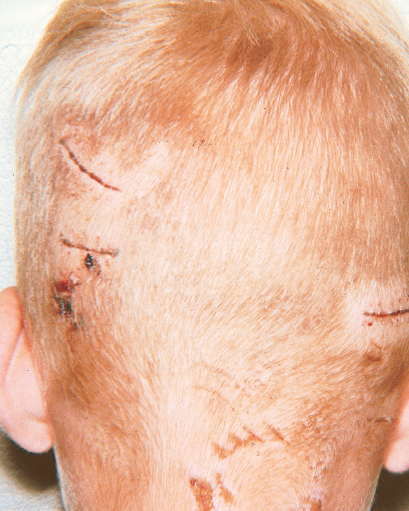
Treatment. The bites were irrigated with saline, and the boy was given an intramuscular injection of ceftriazone. Amoxicillin-clavulanate was prescribed to prevent infection with streptococci, staphylococci, Pasteurella organisms, and anaerobes. A booster dose of tetanus and diphtheria toxoids was given.
Outcome of the case. At reexamination 3 days later, the child was afebrile, there was no purulent discharge from the healing wounds, and he said that he felt fine. Follow-up at 1 week was uneventful. The dog’s owners said the animal had been immunized for rabies.
Discussion. Every year, at least 10 children die of dog bites.1 Most biting dogs are not strays and are known to the victim and his or her family.
Reference:
1. Sacks JJ, Lockwood R, Hornreich J, Sattin RW. Fatal dog attacks, 1989-1994. Pediatrics. 1996;97(6 Pt 1):891-895.