
5 Uncommon Cutaneous and Subcutaneous Malignancies: Identification, Diagnosis, and Treatment
ABSTRACT: The vast majority of skin growths or tumors are benign. It is very common for patients to ask their health care provider about such lesions, and they often seek advice when consulting about some other health problem. However, some of these lesions may be cutaneous and subcutaneous malignancies, which are uncommon and can be difficult to identify. This article discusses signs and symptoms, diagnosis, and treatment options for 5 cutaneous and subcutaneous malignancies that you may encounter in your practice: primary cutaneous lymphomas, Merkel cell carcinoma, Kaposi sarcoma, adnexal tumors, and metastatic cancers to the skin.
KEYWORDS: Primary cutaneous lymphoma, Merkel cell carcinoma, Kaposi sarcoma, adnexal tumor, metastatic cancers to the skin
Cutaneous malignancies vary widely in their aggressiveness, morbidity, and mortality. Basal cell and squamous cell carcinomas are by far the most common cutaneous malignancies. Malignant melanoma is less common and is much more aggressive. This review article covers the recognition, diagnosis, and treatment of less common cutaneous and subcutaneous malignancies. Although they are uncommon, it is important to have the ability to recognize these pathologies. The following malignancies are included: primary cutaneous lymphomas, Merkel cell carcinoma, Kaposi sarcoma, adnexal tumors, and metastatic cancers to the skin.
Primary Cutaneous Lymphomas
Primary cutaneous lymphomas are a heterogeneous group of non-Hodgkin lymphomas that affect the skin and may progress to systemic disease. These have replaced Hodgkin disease as the most common adult lymphomas, and they are more common among the black population than the white population and are more common in men than in women.1
The majority of cases are cutaneous T-cell lymphomas, comprising 65% to 92% of cutaneous lymphomas, of which mycosis fungoides and Sézary syndrome are most common.2 Mycosis fungoides has more of an indolent behavior, and Sézary syndrome frequently is more aggressive. The tumors of mycosis fungoides can become very large and may be mushroom-shaped, thus the term fungoides.1 A smaller subset of cutaneous lymphomas includes primary cutaneous CD4+ small/medium pleomorphic lymphoma, cutaneous γδ T-cell lymphoma, and cutaneous B-cell lymphoma. The T cells affected are those responsible for skin homing.
The cutaneous lymphomas present a diagnostic challenge, because they can mimic several other dermatologic diseases, including psoriasis, contact dermatitis, nummular eczema, atopic dermatitis, lichen simplex chronicus, lymphoid contact dermatitis, and tinea corporis. Differentiation of cutaneous lymphomas and other conditions is done through historical information, clinical presentation, and histopathologic analysis.
Signs and symptoms. Historically, primary cutaneous lymphomas may be present for long periods with an indolent course. Patients complain of itching, scaling lesions that can be a single lesion or multiple lesions, and that can affect the trunk, face, scalp, inner arms, and inner thighs. Clinically, patches of scaling areas can be seen, and no specific pattern is noted (Figure 1). Some lesions may go undiagnosed for years and may respond to topical corticosteroids or resolve spontaneously. These lesions may be erythematous patches or plaques and can be scaly or wrinkled.
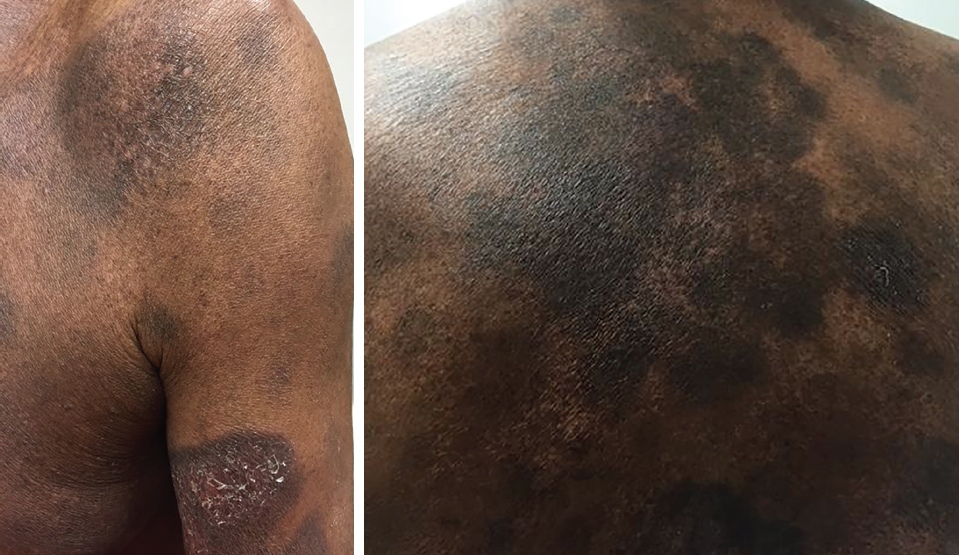
Figure 1. Primary cutaneous lymphoma lesions.
Histology. Histopathologic biopsy of the affected areas shows atypical lymphoid infiltration and may show CD4/CD8 ratio and CCR10 positivity. Biopsy should be taken from different areas after discontinuing treatment for several weeks. The peripheral blood in patients with Sézary syndrome will show Sézary cells, which are atypical hyperconvoluted cerebriform lymphocytes. These findings along with the results of flow cytometry help differentiate cutaneous lymphomas from other skin pathologies and specify the type of cutaneous lymphomas present, as well as assist in guiding treatment.
Staging is based on the tumor-node-metastasis-blood (TNMB) system tumor description, lymph node involvement, the presence of metastasis, and blood involvement. Areas of distant metastasis may involve the gastrointestinal tract, the spleen, or the lungs. These types of cutaneous lymphomas have been classified by the World Health Organization–European Organization for Research and Treatment of Cancer (WHO-EORTC) and is based on clinical and histologic markers.
Treatment. Treatment of cutaneous lymphomas varies widely and is based on the number and size of lesions, TNMB staging, and WHO-EORTC classification. Although most cases are not cured and have a chronic relapsing course, complications can be managed with several modalities.
Treatment goals are clearance of skin disease, minimization of recurrence, prevention of disease progression, and preservation of life.3 Small patch or plaque lesions may be treated with corticosteroids or nitrogen mustard, and more widespread disease is treated with phototherapy as well as low-dose immunomodulators such as interferon.3
In addition to these treatments, other modalities used for this disorder include psoralen–UV-A therapy, radiation therapy, both localized and total skin electron radiation, topical retinoids, bexarotene, biologics (eg, alemtuzumab), oral etretinate, carmustine, histone deacetylase inhibitors, imiquimod, and intralesional injections of interferon.1,4
Merkel Cell Carcinoma
Merkel cell carcinoma (MCC) is a rare and highly aggressive cutaneous neuroendocrine small-cell malignancy. It is highly metastatic to the regional lymphatic basin, as well as nodal and hematogenous spread, and it is often fatal with a 33% mortality rate.1,5 MCC also is known as apudoma, primary neuroendocrine carcinoma of the skin, primary small cell carcinoma of the skin, and trabecular carcinoma of the skin.6 The etiology includes UV solar radiation exposure, immunosuppression, advanced age, and Merkel cell polyomavirus (MCPyV).7-11
Polyomaviruses are nonenveloped, double-stranded DNA viruses that are common in humans. All polyomaviruses encode the large T-antigen and small t-antigen proteins. At present, 13 different human polyomaviruses are known. Some of the large T-antigen and small t-antigen viruses have been shown to possess oncogenic properties.12 In 2008, MCPyV was newly identified and appears to be the only human polyomavirus associated with cancer.10,12 MCPyV can be acquired through close contact involving respiratory tract secretions and can cause widespread, previously unrecognized asymptomatic infection in immunocompromised children and adults.13 MCPyV is highly prevalent in the healthy population. The virus has been identified in approximately 80% of MCC tumors.1,11
Signs and symptoms. MCC is most commonly located on sun-exposed skin such as the head, neck, and extremities, and it is more common in white, elderly men older than 65 years.5,7,9 The typical presentation is a painless, rapidly enlarging, dome-shaped, red or purplish nodule (Figure 2). It also may have a plaque-like appearance or a subcutaneous mass without epidermal changes. In the early stages of MCC, the lesions may resemble a benign entity, causing delay in diagnosis. Spontaneous regression of cutaneous primary tumors also may occur. The differential diagnosis may include basal cell carcinoma, cyst, squamous cell carcinoma, pyogenic granuloma, melanoma, lymphoma cutis, or lipoma.8,14

Figure 2. Merkel cell carcinoma on the right eyebrow. (Credit: Open-i Open Access Biomedical Image Search Engine, https://openi.nlm.nih.gov)
Histology. Histologically, the tumor commonly involves the full thickness of the dermis and frequently extends into the subcutaneous fat and adjacent skeletal muscle. MCC often has typical histopathologic and immunohistochemical features. It tends to show classic histologic features of a neuroendocrine carcinoma and often is positive for CK20 or AM5 proteins. Previously unrecognized paranuclear dot-like expression of CD99 may be used in differentiating MCC from other cutaneous malignancies, especially when CK20 expression is limited or absent. MCC forms sheets and cords of small blue cells, typically in the dermis or subcutaneous tissue. Sometimes the diagnosis is missed because of atypical histologic or aberrant immunohistochemical findings.1,14-16
Treatment. MCCs of all sizes have metastatic potential.1 Therefore, early diagnosis is essential to decrease chances of metastasis, since metastatic disease has a poorer prognosis. Pathologic assessment of regional lymph nodes with sentinel lymph node biopsy in patients without clinical involvement has permitted more accurate staging and more appropriate management.5 Surgical excision typically is the treatment of choice, with wide local excision or Mohs surgery and regional radiotherapy.
Studies show that for uncomplicated head and neck MCC, treatment with surgery and adjuvant radiotherapy is effective in increasing survival and reducing recurrence.8,9,17 Radiation therapy often is reserved for cases in which surgery is not the preferred choice or for high-risk cases in which adjuvant therapy is recommended.9 The disease carries an overall 5-year survival rate of approximately 30% to 60%.1,18
Kaposi Sarcoma
Kaposi sarcoma (KS) is a rare disease and generally is not believed to be a true malignancy but rather a virally induced angioproliferative disorder associated with human herpesvirus 8 (HHV-8).19 HHV-8 is required for the development of KS; however, cofactors such as an immunocompromised state also are necessary for KS development. The 4 subtypes of KS are classic, AIDS-associated, endemic, and immunosuppression-associated.
Classic KS is a disease of older men of Eastern European or Mediterranean descent. It was first described by Hungarian dermatologist Moritz Kaposi in 1872.20 Classic KS occurs more commonly in men, with a male to female ratio as high as 15 to 1, and a median age of 64 years.21
In classic KS, skin lesions often appear initially on the lower extremities and may spread centrally. Lesions progress slowly, often over 10 or more years, and the disease has a benign course.
AIDS-associated KS is the most common AIDS-associated malignancy and is currently the most prevalent form of KS.22 AIDS-associated KS tends to be rapidly progressive. In AIDS-associated KS, treatment of AIDS with highly active antiretroviral therapy can significantly improve KS lesions. Specific treatment of KS lesions, however, does not lead to improvement in AIDS.
Endemic KS had been the predominant form of KS in sub-Saharan Africa prior to the AIDS epidemic. Endemic KS is more aggressive than classic KS and can affect the lymphatic system, becoming fatal.23 It is found equally among men and women, and it generally occurs at a younger age than does classic KS. Endemic KS also may affect children, in whom it rapidly progresses with lymphatic involvement.
Immunosuppression-associated KS occurs during a severely immunocompromised state, such as during organ transplantation, autoimmune conditions, or in patients with malignancy. The appearance of iatrogenic KS often occurs several years after receiving immunosuppressive agents, particularly in patients receiving higher doses.24 Lesions can regress when the immunosuppressive medications are reduced or discontinued, and the condition often is more aggressive when higher doses are used.25
Signs and symptoms. KS is a progressive skin condition with ecchymosis-like macules evolving into papules, plaques, and finally purplish brownish nodules (Figure 3). The lesions often begin on the extremities and progress centrally over time. As lesions coalesce, lymphedema of the surrounding tissue can occur. Definitive diagnosis is achieved with skin biopsy.

Figure 3. AIDS-associated Kaposi sarcoma. (Credit: Open-i Open Access Biomedical Image Search Engine, https://openi.nlm.nih.gov)
Visceral or mucosal involvement is found in 10% of patients with classic KS and is more common in AIDS-associated KS.26 Involvement of the mucous membranes of the mouth and gastrointestinal tract, including the liver and spleen, can occur. These lesions generally are asymptomatic but can cause bleeding from the gastrointestinal tract. Regional lymphadenopathy also can occur but is generally rare, with the exception of children with KS and in AIDS-associated KS.
Treatment. There is no curative treatment for KS; therefore, palliative therapy is aimed at improving lymphedema, decreasing the size of skin or visceral lesions, and delaying or preventing disease progression. For patients with slow progression of disease, observation alone may be appropriate. Radiotherapy is very effective for treating KS lesions, although it is limited due to the widespread nature of the disease. Cutaneous KS lesions may also be treated with excision for single lesions, cryotherapy for small lesions, and intralesional therapy and topical therapy.
Intralesional injections of chemotherapy utilize vinblastine or bleomycin, whereas topical therapies have included imiquimod and cis-retinoic acid.27 Chemotherapy and/or immunomodulators may be used when there is significant disease burden, very aggressive progression, or significant visceral involvement. Several chemotherapy agents are approved by the US Food and Drug Administration for AIDS-associated KS, including pegylated liposomal doxorubicin, vinblastine, and paclitaxel.28 Pegylated liposomal doxorubicin is considered first-line therapy for AIDS-associated KS and often is used off-label for classic KS, as well.29 Recombinant interferon-alfa and thalidomide have anti-angiogenic and immunomodulatory properties and are approved treatments for AIDS-associated KS but have limited use in classic and endemic KS.30
Apocrine and Adnexal Carcinomas
Apocrine and adnexal carcinomas are endocrine mucin-producing tumors that are rare and very commonly are misdiagnosed. This is due to the general infrequency of each of these conditions, as well as similar features to more common skin conditions. Apocrine carcinomas occur at an extremely rare rate of less than 0.005% of all tumors resected.31 Adnexal carcinomas are slightly more common (1% to 2% of skin cancers).32 Both pose a diagnostic challenge because of their benign appearance.
Signs and symptoms. Adnexal carcinomas are commonly found on the head and neck and less frequently on the extremities and trunk. They may arise from multiple sites such as apocrine and eccrine sweat glands, hair follicles, and sebaceous glands. Because of the lack of information about and rarity of adnexal tumors, one may not confidently identify a specific age group or race that is susceptible to them. Adnexal masses have a hard texture and, depending on the location, may have different appearances such as thickened skin; red, white, blue or black pigments; excrescence; and cystic or solid features (Figure 4). These tumors minimally change the skin, begin as a single “bump,” and are round or ovular. This benign appearance may lead to the tumor advancing to a larger size before surgical excision is performed. Development of these tumors typically is slow, and metastasis is rare.

Figure 4. Adnexal carcinoma (specifically, trichilemmal carcinoma).
Apocrine carcinoma is found most often in areas of high apocrine sweat gland density such as the axilla, ear, eyelid, areola, and perineum. Rather than opening to the surface of the skin, sweat is secreted into the pilary canal of the hair follicle.31 Apocrine carcinomas are characterized by their nontender, firm, or rubbery masses that are found subcutaneously and intradermally (Figure 5). The overlying skin takes on a red or purple hue, and is indurated.

Figure 5. Apocrine carcinoma (specifically, apocrine papillated cystadenoma). (Credit: Derm101, http://www.derm101.com/clinical-atlas/miscellaneous-benign-neoplasms)
Histology. The diagnosis of adnexal carcinomas is made by biopsy and immunohistochemical studies and histologic features. Immunohistochemical reports of GATA3 have been cited in benign and malignant epidermal cutaneous adnexal neoplasms.33 Diagnosis of apocrine carcinomas is based on biopsy with immunohistochemistry and ultrastructural features. They are easier to identify than most adnexal carcinomas due to the location, prominent features, appearance on sweat gland, and changes of the skin pigmentation around them. Tumor cells are positive on periodic acid–Schiff staining due to glycogen granules.31 Despite these identifying characteristics, the variability of histologic features has led to some confusion regarding the classification. Due to the large number of these rare entities, a multiplicity of names to designate the same neoplasm, and a lack of consensus regarding classification and nomenclature, these tumors may present a diagnostic challenge.34
Treatment. Apocrine and adnexal carcinomas are treated in the same manner. Wide surgical excision of the carcinoma along with regional lymph node dissection is the recommended treatment. Frequent follow-ups are essential to detect recurrence and metastasis.31 Many of the adnexal and apocrine carcinomas are chemotherapy- and radiation-resistant; therefore, these rarely are viable options.
Metastatic Cancers to Skin
Cutaneous metastasis of cancers originating in other organs can occur by hematologic or lymphatic embolization, or direct implantation during surgical procedures.35
Many primary tumors have the potential to metastasize to the skin and subcutaneous tissues. These include breast cancer most commonly, but it also can occur with esophageal, gastric, or colon cancers, nasopharyngeal cancer, lymphomas, pancreatic cancer, renal cell carcinoma, lung cancer, and ovarian cancer.
Signs and symptoms. The cutaneous and subcutaneous lesions can be the presenting sign of distant metastasis, or they can be found in patients with known primary cancers. Skin and subcutaneous metastasis of primary distant tumors carries a poorer prognosis. Location of cutaneous metastasis varies and can include the chest, pelvis, face, and scalp but may occur anywhere on the body. Metastatic cutaneous and subcutaneous skin lesions may have varying appearance. They may appear as painless, flesh-colored nodules and may be pigmented or friable skin neoplasms. Lesions may be single or multiple with widespread coverage (Figure 6).
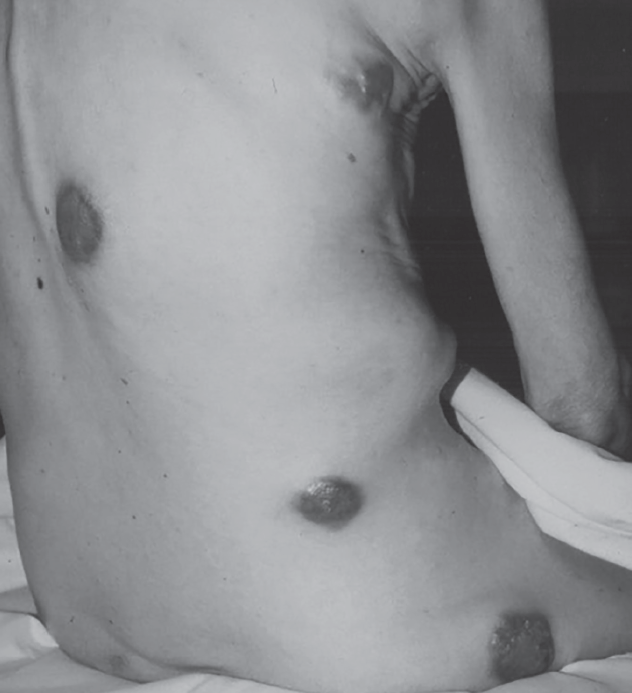
Figure 6. Diffuse cutaneous metastases from colon cancer. (Credit: Open-i Open Access Biomedical Image Search Engine, https://openi.nlm.nih.gov)
Histology. Diagnosis of these neoplasms is made by biopsy with microscopic and immunohistochemical analysis.
Treatment. Treatment depends on the primary source of the malignancy and may include systemic chemotherapy and local external beam radiation treatments to treat the individual metastatic cutaneous lesions.
Charles J. Haddad, MD, is an associate professor at the University of Florida in Jacksonville.
Judella Haddad-Lacle, MD, is an associate professor at the University of Florida in Jacksonville.
Reetu Grewal, MD, is an assistant professor at the University of Florida in Jacksonville.
Lori Bilello, PhD, is an assistant professor at the University of Florida in Jacksonville.
Charles M. Haddad is a student at Florida State University in Tallahassee.
References:
- Habif TP. Premalignant and malignant nonmelanoma skin tumors. In: Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 6th ed. Philadelphia, PA: Elsevier; 2016:809-852.
- Ally MS, Robson A. A review of the solitary cutaneous T-cell lumphomas. J Cutan Pathol. 2014; 41(9):703-714.
- Jawed SI, Myskowski PL, Horwitz S, Moskowitz A, Querfeld C. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part II: prognosis, management, and future directions. J Am Acad Dermatol. 2014; 70(2); 223.e1-223.e17.
- Miyagaki T, Sugaya M. Erythrodermic cutaneous T-cell lymphoma: how to differentiate this rare disease from atopic dermatitis. J Dermatol Sci. 2011;64(1):1-6.
- Gunaratne DA, Howle JR, Veness MJ. Sentinel lymph node biopsy in Merkel cell carcinoma: a 15-year institutional experience and statistical analysis of 721 reported cases. Br J Dermatol. 2016;174(2):273-281.
- Argenyi ZB. Neural and neuroendocrine neoplasms (other than neurofibromatosis). In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 1. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012:1943-1960.
- Lowell DL, Roberts J, Gogate P, Goodwin R. Merkel cell carcinoma: case study and literature review. J Foot Ankle Surg. 2014;53(2):219-225.
- Saini AT, Miles BA. Merkel cell carcinoma of the head and neck: pathogenesis, current and emerging treatment options. Onco Targets Ther. 2015;8:2157-2167.
- Porceddu SV, Veness MJ, Guminski A. Nonmelanoma cutaneous head and neck cancer and Merkel cell carcinoma: current concepts, advances, and controversies. J Clin Oncol. 2015; 33(29):3338-3345.
- Samimi M, Gardair C, Nicol JT, Arnold F, Touzé A, Coursaget P. Merkel cell polyomavirus in Merkel cell carcinoma: clinical and therapeutic perspectives. Semin Oncol. 2015;42(2):347-358.
- Grundoff A, Fischer N. Merkel cell polyomavirus, a highly prevalent virus with tumorigenic potential. Curr Opin Virol. 2015;14:129-137.
- Moens U, Rasheed K, Abdulsalam I, Sveinbjørnsson B. The role of Merkel cell polyomavirus and other human polyomaviruses in emerging hallmarks of cancer. Viruses. 2015;7(4): 1871-1901.
- Sourvinos G, Mamma IN, Spandidos DA. Merkel cell polyomavirus infection in childhood: current advances and perspectives. Arch Virol. 2015; 160(4):887-892.
- Munde PB, Khandekar SP, Dive AM, Sharma A. Pathophysiology of Merkel cell. J Oral Maxillofac Pathol. 2013;17(3):408-412.
- Goessling W, McKee PH, Mayer RJ. Merkel cell carcinoma. J Clin Oncol. 2002;20(2):588-598.
- Succaria F, Radfar A, Bhawan J. Merkel cell carcinoma (primary neuroendocrine carcinoma of skin) mimicking basal cell carcinoma with review of different histopathologic features. Am J Dermatopathol. 2014;36(2):160-166.
- Raju S, Vazirnia A, Totri C, Hata TR. Treatment of Merkel cell carcinoma of the head and neck: a systematic review. Dermatol Surg. 2014; 40(12):1273-1283.
- Schrama D, Ugurel S, Becker JC. Merkel cell carcinoma: recent insights and new treatment options. Curr Opin Oncol. 2012;24(2):141-149.
- Geraminejad P, Memar O, Aronson I, Rady PL, Hengge U, Tyring SK. Kaposi’s sarcoma and other manifestations of human herpesvirus 8. J Am Acad Dermatol. 2002;47(5):641-655.
- Kaposi M. Idiopathic multiple pigmented sarcoma of the skin. CA Cancer J Clin. 1982;32(6): 342-347.
- Antman K, Chang Y. Kaposi’s sarcoma. N Engl J Med. 2000;342(14):1027-1038.
- Feigal EG. AIDS-associated malignancies: research perspectives. Biochim Biophys Acta. 1999;1423(1):C1-C9.
- Wabinga HR, Parkin DM, Wabwire-Mangen F, Mugerwa JW. Cancer in Kampala, Uganda, in 1989-91: changes in incidence in the era of AIDS. Int J Cancer. 1993;54(1):26-36.
- Cathomas G, Tamm M, McGandy CE, et al. Transplantation-associated malignancies: restriction of human herpes virus 8 to Kaposi’s sarcoma. Transplantation. 1997;64(1):175-178.
- Trattner A, Hodak E, David M, Sandbank M. The appearance of Kaposi sarcoma during corticosteroid therapy. Cancer. 1993;72(5):1779-1783.
- Rappersberger K, Stingl G, Wolff K. Kaposi’s sarcoma. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Fitzpatrick’s Dermatology in General Medicine. Vol 1. 5th ed. New York, NY: McGraw-Hill; 1999:1195-1204.
- Krown, SE, Singh JC. Classic Kaposi sarcoma: clinical features, staging, diagnosis, and treatment. UpToDate. http://www.uptodate.com/contents/classic-kaposi-sarcoma-clinical-features-staging-diagnosis-and-treatment. Updated July 10, 2014. Accessed May 10, 2016.
- Gill PS, Rarick M, McCutchan JA, et al. Systemic treatment of AIDS-related Kaposi’s sarcoma: results of a randomized trial. Am J Med. 1991; 90(4):427-433.
- Di Lorenzo G, Kreuter A, Di Trolio R, et al. Activity and safety of pegylated liposomal doxorubicin as first-line therapy in the treatment of non-visceral classic Kaposi’s sarcoma: a multicenter study. J Invest Dermatol. 2008;128(6):1578-1580.
- Krown SE. Management of Kaposi sarcoma: the role of interferon and thalidomide. Curr Opin Oncol. 2001;13(5):374-381.
- Kshirsagar AY, Wader JV, Nagur B, et al. Case report: a rare case of eccrine carcinoma. Int J Surg Case Rep. 2015;15:149-151.
- Guerrissi JO, Quiroga JP. Adnexal carcinomas of the head and neck. Indian J Plast Surg. 2008; 41(2):229-234.
- Mertens RB, de Peralta-Venturina MN, Balzer BL, Frishberg DP. GATA3 expression in normal skin and in benign and malignant epidermal and cutaneous adnexal neoplasms. Am J Dermatopathol. 2015;37(12):885-891.
- Cardoso JC, Calonje E. Malignant sweat gland tumours: an update. Histopathology. 2015;67(5): 589-606.
- Bittencourt MdJS, Carvalho AH, Nascimento BAMd, Freitas LKM, Parijós AMd. Cutaneous metastasis of a breast cancer diagnosed 13 years before. An Bras Dermatol. 2015; 90(3 suppl 1):134-137.