
Teenager With Recalcitrant Rash and Lower Extremity Pain

A 16-year-old girl presented with a rash of 4 months’ duration. Initially, the rash consisted of pruritic, erythematous macules with excoriations, but no pustules or drainage, located on the right shin.
A similar rash occurred 1 week later on the left shin. There was no history of insect bites or atopy.
The rash significantly improved following twice-daily application of 0.1% triamcinolone cream. Treatment with bacitracin for suspected superimposed bacterial infection worsened the rash. Oral dicloxacillin resulted in some improvement.
One month after she initially presented with the rash, the patient was seen for a soccer preparticipation physical examination. At this time the rash was no longer pruritic but remained otherwise essentially unchanged. Purpuric dermatosis was suspected, and the patient was referred to a dermatologist. A complete blood cell count was done to rule out infection as part of a work-up for suspected vasculitis; it was essentially normal except for a mild basophilia.
Seven months after the onset of symptoms, the patient was seen again for the rash. The dermatologist had initially included erythema annulare centrifugum and tinea in the differential diagnosis, and had treated the patient with sequential courses of itraconazole and ciclopirox. The rash improved significantly and appeared only as fading annular red patches. Given the patient’s initially favorable response to topical corticosteroids, a 3-week course of halobetasol was prescribed.
More than a year later, the rash was still present (Figure 1). The patient’s primary care physician obtained a 4-mm punch biopsy from the anterior portion of the left shin, and the site was sutured with adequate healing. Biopsy revealed vasculopathy with erythrocyte extravasation (purpura). Hyaline thrombi were seen in blood vessels, possibly representing fibrin or other substance (cryglobulin). These histologic changes may be seen in several disorders, including livedoid vasculitis, cryoglobulinemia, hypercoagulable states, and antiphospholipid syndromes, as well as pigmented purpuric dermatoses. A general lack of inflammation excluded leukocytoclastic vasculitis.
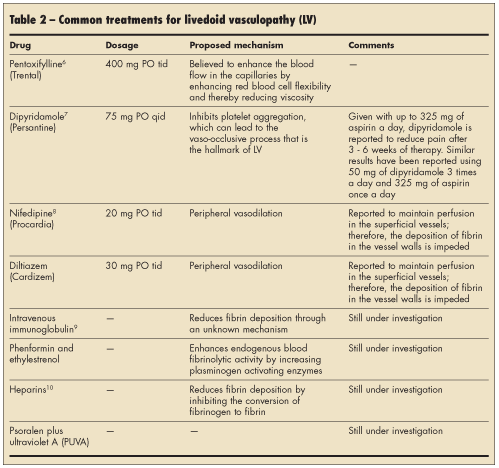
Two months later, the patient presented with right ankle pain, and she reported that the rash had become painful. Blood tests, including tests for hypercoagulable disorders, revealed only mild basophilia without other significant abnormalities. Livedoid vasculopathy was diagnosed. Oral pentoxifylline 400 mg twice daily was started and subsequently increased to 3 times a day.
Three years after the rash first appeared, the patient presented with left lower leg pain of rapid onset, sharp quality, and moderate to severe intensity; it worsened with weight bearing and there was no radiation. She reported significantly increased periods of weight bearing at work.
Examination revealed a few resolving ulcerations with marginal erythema, minimal warmth, and exquisite tenderness on the lower third of the left anterior shin. Distal pulses were 21 equal bilaterally. The results of the remainder of the musculoskeletal examination were grossly normal.
She was started on oral ibuprofen 600 mg every 6 hours as needed for pain with hydrocodone/acetaminophen to be used for breakthrough pain. She was encouraged to continue to take pentoxifylline 3 times a day. Results of blood tests for prothrombin 20210 gene mutation and anti-dsDNA and hepatitis screening were all negative. The patient has experienced significant improvement in symptoms with pentoxifylline and aspirin taken as needed for pain.
LIVEDOID VASCULOPATHY: AN OVERVIEW
Livedoid vasculopathy is characterized by ulceration of the lower extremities, and it can evolve into a dermatological finding termed atrophie blanche, which consists of white stellate stars. Unlike the small-vessel vasculitides (eg, microscopic polyangiitis, Churg-Strauss syndrome, and drug-induced vasculitis), livedoid vasculopathy is a distinct condition that is not usually the result of other diseases. Biopsy can aid in making the diagnosis, but histologic findings are not pathognomonic. The condition manifests with segmental hyalinizing vascular involvement of thickened dermal blood vessels, endothelial proliferation, and focal thrombosis without nuclear dust. Direct immunofluorescence study reveals immunoglobulin and complement components in the superficial, mid-dermal, and deep dermal vessels.
ETIOLOGY AND PATHOPHYSIOLOGY
The etiology of livedoid vasculopathy is uncertain. The histology evolves according to the temporal stage of the lesion. Most commonly, fibrin deposition is found within both the wall and the lumen of affected vessels.1 The absence of a substantial perivascular infiltrate or leukocytoclasia argues against a vasculitis; it is more in keeping with a thrombo-occlusive process.
The underlying mechanism of the development of livedoid vasculopathy may be related to the development of a fibrin cuff, white-cell trapping, microthrombi, a defect of endothelial cell plasminogen activator, platelet dysfunction, and enhanced fibrin formation. The following is a summary of the excellent discussion of these mechanisms by Maessen-Visch and associates2 in 1999.
In their fibrin cuff theory, Browse and Burnand3 postulate that because of chronic venous compromise, a leakage of fibrinogen from the capillaries induces a fibrin cuff around the capillaries, leading to a barrier for oxygen and nutrients. In 1988, Coleridge-Smith and colleagues4 suggested the white-cell trapping theory. White cells adhere (trap) to the endothelium of the capillaries as a result of venous hypertension. This results in the induction of proteolytic enzymes and superoxide metabolites, causing destruction of the tissue. This occurrence appears to be a nonimmunologic phenomenon; it results from the low flow in the wide capillaries.
In their research, Klein and Pittelkow5 noted that a defect of endothelial cell plasminogen activator exists in some patients with livedoid vasculopathy, with a high incidence of impaired release of tissue plasminogen activator (tPA), increased levels of tPA inhibitor, and a markedly increased incidence of antiphospholipid antibodies. In other studies, levels of tPA in the reference range were found in patients with chronic venous insufficiency and atrophie blanche or lipodermatosclerosis. Other evidence has implicated increased platelet aggregation in patients with livedoid vasculopathy.
SIGNS AND SYMPTOMS
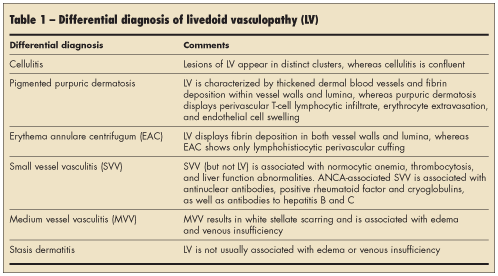
A thorough history should be obtained to rule out more common conditions (Table 1). In livedoid vasculopathy, the initial findings are typically painful, purpuric macules or papules on the ankles and the adjacent dorsum of the feet.1 Patients may have a history of livedo reticularis on their lower legs.
Medium-sized vasculitides, such as polyarteritis nodosa (PAN), occasionally present with ulceration, resulting in ivory-white, stellate scarring on the lower limbs. These conditions may potentially be misdiagnosed as livedoid vasculopathy. Furthermore, livedoid vasculopathy is not associated with edema or venous insufficiency, whereas stasis dermatitis with ulceration is usually not painful and is associated with obvious edema and signs of venous insufficiency.
Patients with livedoid vasculopathy may also have a history of recurrent leg ulcerations. Such patients can have deficiencies in a variety of blood factors (eg, factor V Leiden, protein C). The factor V Leiden mutation is more frequent in patients with venous leg ulceration than in control subjects and the general population. Patients with the factor V Leiden mutation are at increased risk for the development of deep venous thrombosis and recurrent leg ulceration. Patients may also have a history of increased plasma homocysteine levels, abnormalities in fibrinolysis, and increased platelet activation.
Finally, livedoid vasculopathy is not as painful as hypertensive ischemic ulcers, which are usually larger and lack telangiectatic purple borders. The history and careful follow-up care can rule out traumatic ulcers and distinguish them from livedoid vasculopathy. Chronic PAN may be associated with painful ulcerations; however, the associated nodules should differentiate chronic PAN from livedoid vasculopathy.
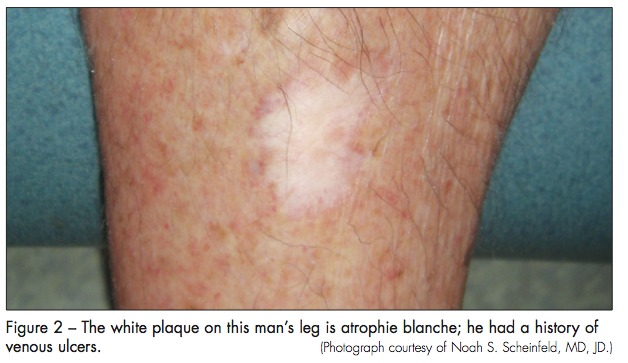
The initial lesions of livedoid vasculopathy, which often appear in clusters or groups, eventually ulcerate over a period of months to years and form irregular patterns of superficial ulcers. When the ulcers finally heal, they leave behind atrophic porcelain-white scars, which are atrophie blanche. Venous stasis and other conditions, such as leukocytoclastic vasculitis, can lead to identical white scars on the legs (Figure 2). Patients who have systemic diseases, such as lupus, rheumatoid arthritis, and Klinefelter syndrome, that can result in skin ulcers may also exhibit atrophie blanche–like lesions. These patients do not have livedoid vasculopathy.
Patients with livedoid vasculopathy may exhibit livedo reticularis before ulceration, with ulcerations, or with no ulcerations. They may have Raynaud phenomenon and acrocyanosis. Furthermore, lipodermatosclerosis can be associated with livedoid vasculopathy.
TREATMENT
While ruling out the various disease states that have been associated with livedoid vasculopathy, physicians can offer a number of therapies that have been very helpful in reducing pain and ulceration. Instituting treatment as soon as possible is best. Various treatments have been proposed, with varying degrees of success from patient to patient, even with similar disease presentation and course. Table 2 summarizes the most commonly used treatment regimens.
Although painful, livedoid vasculopathy is not associated with loss of life or limb. While it is also mostly a disease of young adults, this case illustrates the importance of early diagnosis and a good therapeutic alliance in order to cope successfully with the potentially devastating emotional scarring in this self-conscious patient population. The patient in this case had resorted to wearing exclusively jeans and long pants even in the midst of summer to avoid embarrassment. Patients must understand that livedoid vasculopathy is a chronic condition whose effects can sometimes be improved by medications.
1. Cocuroccia B, Tonanzi T, Menaguale G, et al. Livedoid vasculopathy and skin ulcers in patients with inherited thrombophilia. Eur J Dermatol. 2002;12(4):360-363.
2. Maessen-Visch MB, Hamulyak K, Tazelaar DJ, et al. The prevalence of factor V Leiden mutation in patients with leg ulcers and venous insufficiency. Arch Dermatol.1999;135(1):41-44.
3. Browse NL, Burnand KG. The cause of venous ulceration. Lancet. 1982;ii:243-245.
4. Coleridge-Smith PD, Thomas P, Scurr JH, et al. Causes of venous ulceration: a new hypothesis. BMJ. 1988;296:1726-1727.
5. Klein KL, Pittelkow MR. Tissue plasminogen activator for treatment of livedoid vasculitis. Mayo Clin Proc. 1992;67(10):923-33.
6. Sams WM. Livedo vasculitis. Therapy with pentoxifylline. Arch Dermatol. 1988;124(5):684-687.
7. Scheinfeld NS. Livedoid vasculopathy. eMedicine World Medical Library. Updated August 1. 2011. Accessed May 17, 2012.
8. Purcell SM, Hayes TJ. Nifedipine treatment of idiopathic atrophie blanche. J Am Acad Dermatol. 1986;14(5 Pt 1):851-854.
9. Amital H, Levy Y, Shoenfeld Y. Use of intravenous immunoglobulin in livedo vasculitis. Clin Exp Rheumatol. 2000;18(3):404-406.
10. Hairston BR, Davis MD, Gibson LE, Drage LA. Treatment of livedoid vasculopathy with low-molecular-weight heparin: report of 2 cases. Arch Dermatol.2003;139(8):987-990.